Mitochondrium jest jednym z najbardziej fascynujących elementów komórki, łącząc w sobie cechy miniaturowej elektrowni, dawnego organizmu bakteryjnego oraz precyzyjnego centrum regulacji procesów życiowych. Zrozumienie budowy i funkcji mitochondriów pozwala wniknąć w sam rdzeń tego, czym jest życie na poziomie molekularnym: skąd bierze się energia, jak powstaje ciepło, w jaki sposób komórki regulują swój los, a organizmy starzeją się lub chorują. To właśnie w mitochondrium splatają się ze sobą metabolizm, genetyka i ewolucja.
Budowa mitochondrium i jego wyjątkowe pochodzenie
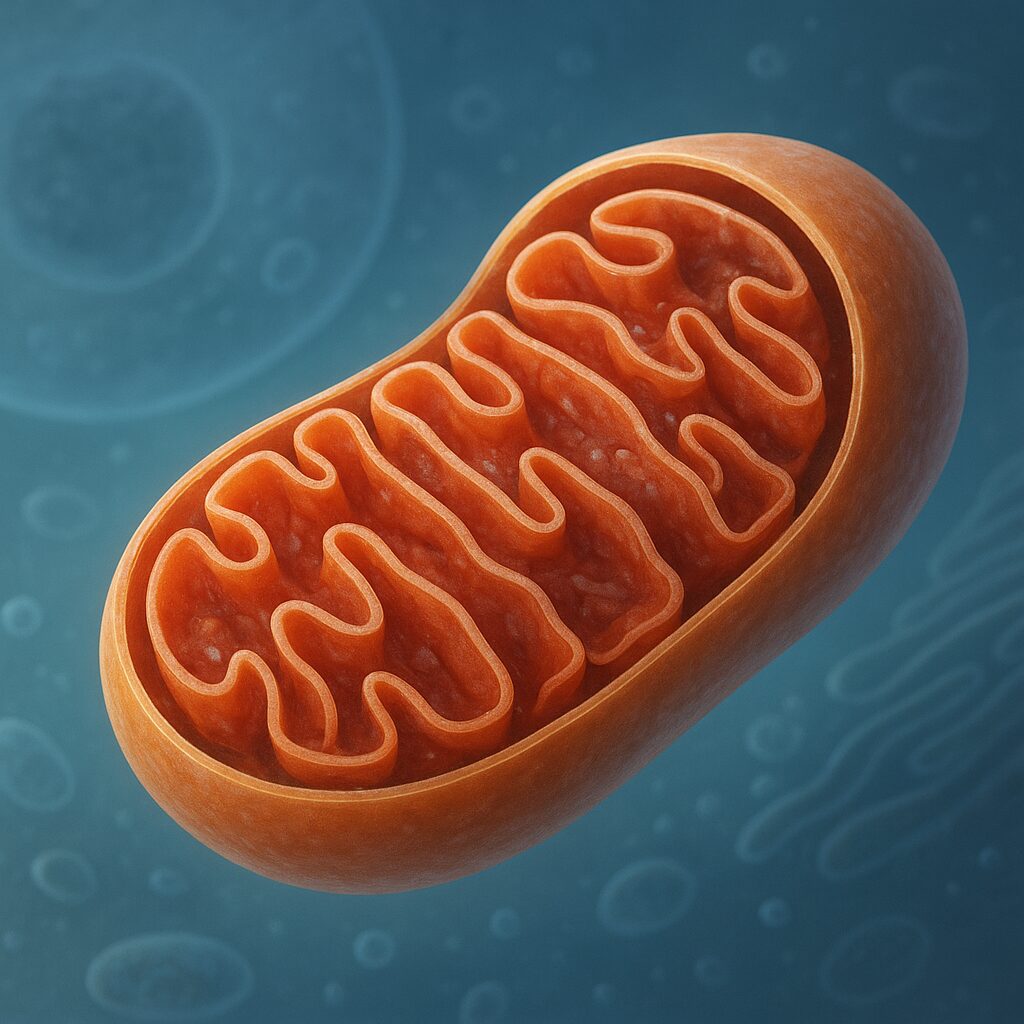
Mitochondria to organella komórkowe otoczone podwójną błoną, występujące niemal we wszystkich komórkach eukariotycznych. Ich struktura jest wysoce wyspecjalizowana, co pozwala prowadzić złożone procesy biochemiczne. Wyróżniamy błonę zewnętrzną, przestrzeń międzybłonową, błonę wewnętrzną oraz macierz mitochondrialną. Taki układ przedziałów umożliwia precyzyjne rozmieszczenie enzymów i kontrolę przepływu jonów oraz metabolitów.
Błona zewnętrzna jest stosunkowo przepuszczalna dla małych cząsteczek dzięki obecności białek kanałowych, tzw. porin. Tworzy ona coś w rodzaju ochronnej osłony, ale jednocześnie nie stanowi poważnej bariery dla wielu metabolitów. Znacznie ważniejsza funkcjonalnie jest błona wewnętrzna, która zawiera liczne białka transportujące, kompleksy enzymatyczne i kanały jonowe. Jest ona silnie pofałdowana, tworząc charakterystyczne grzebienie, czyli kryszty. Zwiększają one powierzchnię roboczą, co ma ogromne znaczenie dla produkcji energii.
Macierz mitochondrialna to żelowa substancja wypełniająca wnętrze organellum. Znajdują się w niej liczne enzymy cyklu kwasu cytrynowego, cząsteczki NAD+ i FAD, rybosomy, a także własne mitochondrialne DNA. Ten ostatni element jest jednym z kluczowych dowodów na teorię endosymbiozy. Zgodnie z nią mitochondria wywodzą się od dawnych bakterii tlenowych, które zostały wchłonięte przez pierwotne komórki eukariotyczne. Z czasem doszło do ścisłej współpracy oraz transferu wielu genów do jądra komórkowego, ale część materiału genetycznego pozostała w organellach.
Teoria endosymbiozy tłumaczy również obecność podwójnej błony: pierwotna błona bakterii stała się dzisiejszą błoną wewnętrzną mitochondrium, a błona gospodarza – błoną zewnętrzną. Dodatkowo, mitochondria dzielą się w sposób przypominający podział bakterii, a ich rybosomy bardziej przypominają rybosomy prokariotyczne niż eukariotyczne. Wszystko to wspiera pogląd, że mitochondria są potomkami dawno zasymilowanych organizmów, które wyspecjalizowały się w wydajnym wykorzystaniu tlenu.
W komórce liczba mitochondriów jest zmienna i zależy od zapotrzebowania energetycznego. Komórki mięśniowe, szczególnie mięsień sercowy, mogą zawierać ich tysiące, podczas gdy komórki o mniejszej aktywności metabolicznej mają ich znacznie mniej. Mitochondria nie są strukturami statycznymi – łączą się, dzielą, przemieszczają po cytoplazmie i podlegają ciągłej wymianie składników, co pozwala na dostosowanie się do bieżących potrzeb komórki.
Rola mitochondrium w produkcji energii i metabolizmie
Najbardziej znaną funkcją mitochondriów jest wytwarzanie energii w postaci cząsteczek ATP, głównego energetycznego „pieniądza” komórki. Proces ten odbywa się głównie poprzez fosforylację oksydacyjną, która łączy szereg reakcji biochemicznych: utlenianie substratów w cyklu kwasu cytrynowego, transport elektronów w łańcuchu oddechowym oraz syntezę ATP przez syntazę ATP. Kluczem jest tworzenie gradientu protonowego między macierzą a przestrzenią międzybłonową.
Cykl kwasu cytrynowego, zwany również cyklem Krebsa, zachodzi w macierzy mitochondrium. W jego trakcie cząsteczki acetylo-CoA, pochodzące głównie z rozkładu glukozy i kwasów tłuszczowych, są całkowicie utleniane do dwutlenku węgla. Jednocześnie powstają zredukowane przenośniki elektronów: NADH i FADH2. To one stanowią paliwo dla łańcucha transportu elektronów, zlokalizowanego w błonie wewnętrznej.
Łańcuch oddechowy składa się z kilku kompleksów enzymatycznych, które przekazują elektrony na tlen cząsteczkowy. W miarę przepływu elektronów energia jest wykorzystywana do pompowania protonów z macierzy do przestrzeni międzybłonowej, tworząc różnicę stężeń i potencjału elektrycznego. Ten gradient protonowy jest rodzajem magazynu energii, który zostaje spożytkowany przez syntazę ATP. Protony wracają do macierzy przez kanał w tym białku, a ich ruch napędza fosforylację ADP do ATP.
Oprócz pełnienia funkcji „elektrowni”, mitochondria są ważnymi ośrodkami metabolizmu tłuszczów. Wewnątrz macierzy zachodzi beta-oksydacja kwasów tłuszczowych, czyli etapowe skracanie ich łańcuchów do cząsteczek acetylo-CoA. Proces ten jest kluczowy dla komórek, które w dużej mierze polegają na lipidach jako źródle energii, na przykład dla mięśnia sercowego czy mięśni szkieletowych podczas długotrwałego wysiłku.
Mitochondria uczestniczą także w metabolizmie aminokwasów i częściowo w glukoneogenezie, czyli procesie tworzenia glukozy z niecukrowych substratów. Ponadto są zaangażowane w utrzymanie równowagi redoks i regulację stężenia jonów wapnia. Wapń, wnikając do mitochondriów, nie tylko wpływa na aktywność niektórych enzymów cyklu kwasu cytrynowego, lecz także moduluje inne sygnały wewnątrzkomórkowe.
Ciekawym zjawiskiem jest sprzężenie między produkcją energii a wytwarzaniem ciepła. W niektórych tkankach, zwłaszcza w brunatnej tkance tłuszczowej, obecne są białka rozprzęgające fosforylację oksydacyjną. Zamiast produkować ATP, wykorzystują one gradient protonowy do generowania ciepła. To właśnie dzięki takim mechanizmom ssaki mogą utrzymywać stałą temperaturę ciała w chłodnym środowisku. Mitochondria stają się wówczas nie tyle elektrownią, co wysoko wyspecjalizowanym „grzejnikiem” biologicznym.
Warto podkreślić, że bilans energetyczny komórki zależy nie tylko od samych mitochondriów, ale i od ścisłej współpracy pomiędzy metabolizmem cytoplazmatycznym a mitochondrialnym. Glikoliza w cytoplazmie dostarcza pirogronian, który wnika do mitochondrium i ulega przekształceniu do acetylo-CoA. W okresach ograniczonego dostępu do tlenu część komórek redukuje aktywność łańcucha oddechowego i przechodzi na mniej wydajny, ale niezależny od tlenu metabolizm beztlenowy. Równowaga między tymi szlakami jest sterowana przez liczne sensory energetyczne i sygnały hormonalne.
Mitochondria w regulacji śmierci komórki i sygnalizacji
Rola mitochondriów nie ogranicza się do dostarczania energii. Organella te są kluczowymi regulatorami śmierci komórki, zwłaszcza w procesie apoptozy. Apoptoza to zaprogramowana, uporządkowana forma śmierci komórkowej, niezbędna dla rozwoju organizmu, utrzymania homeostazy tkanek i eliminacji komórek uszkodzonych lub potencjalnie niebezpiecznych. Mitochondria kontrolują uwalnianie białek, które inicjują kaskadę zdarzeń prowadzących do rozpadu komórki.
W warunkach stresu komórkowego, np. w odpowiedzi na uszkodzenia DNA, niedobór składników odżywczych lub działanie toksyn, dochodzi do zmian przepuszczalności błony wewnętrznej i zewnętrznej mitochondrium. Jednym z kluczowych białek jest cytochrom c, który w normalnych warunkach uczestniczy w łańcuchu oddechowym. Gdy zostanie uwolniony do cytoplazmy, wiąże się z innymi składnikami i tworzy kompleks aktywujący kaspazy – proteazy odpowiedzialne za demontaż struktury komórkowej.
Błona zewnętrzna mitochondrium jest regulowana przez liczną rodzinę białek Bcl-2, z których część działa proapoptotycznie, a część antyapoptotycznie. To dynamiczna równowaga między sygnałami „życia” i „śmierci” decyduje o tym, czy komórka uruchomi program apoptozy. Mitochondria są więc swoistym centrum decyzyjnym: reagują na bodźce pochodzące z jądra, cytoplazmy i środowiska zewnętrznego, integrują je i przekuwają w ostateczną odpowiedź.
Poza apoptozą mitochondria uczestniczą w innych formach śmierci komórkowej, w tym w nekroptozie i ferroptozie, choć ich rola jest tu bardziej złożona i nie zawsze bezpośrednia. Co więcej, organella te są włączone w rozbudowaną sieć szlaków sygnalizacyjnych, w której biorą udział reaktywne formy tlenu (ROS). ROS, wytwarzane w niewielkich ilościach podczas pracy łańcucha oddechowego, mogą pełnić rolę cząsteczek sygnałowych, modulując aktywność białek i ekspresję genów.
Nadmierna produkcja ROS prowadzi jednak do stresu oksydacyjnego, uszkodzeń białek, lipidów i kwasów nukleinowych. Mitochondrialne DNA jest szczególnie narażone na tego rodzaju uszkodzenia, ponieważ znajduje się blisko źródła ROS i jest słabiej chronione niż genom jądrowy. Kumulacja mutacji w mitochondrialnym DNA może zaburzać funkcjonowanie łańcucha oddechowego, pogłębiać powstawanie ROS i w efekcie tworzyć błędne koło uszkodzeń.
Reaktywne formy tlenu stanowią jednak także ważny sygnał adaptacyjny. Umiarkowany wzrost ich poziomu może pobudzać mechanizmy obronne komórki, zwiększać ekspresję enzymów antyoksydacyjnych oraz stymulować procesy naprawcze. W ten sposób mitochondria pośrednio wpływają na zdolność komórek do przystosowania się do zmiennych warunków, np. ograniczenia dostępu tlenu czy składników odżywczych.
W kontekście sygnalizacji nie można pominąć roli mitochondriów w regulacji stężenia wapnia. Jony Ca2+ wnikają do mitochondriów przez specjalne kanały i są tam przechowywane lub uwalniane w zależności od potrzeb. Wapń wpływa na aktywność licznych enzymów mitochondrialnych, ale także na procesy w cytoplazmie i jądrze. Dysfunkcja w gospodarce wapniowej może prowadzić do nadmiernej aktywacji enzymów degradacyjnych oraz nasilenia procesów śmierci komórkowej.
Mitochondrialne DNA, dziedziczenie i choroby
Jedną z najbardziej wyjątkowych cech mitochondriów jest posiadanie własnego genomu. DNA mitochondrialne ma postać najczęściej kolistej cząsteczki, zlokalizowanej w macierzy. Zawiera relatywnie niewielką liczbę genów, kodujących część białek łańcucha oddechowego, tRNA i rRNA. Zdecydowana większość białek mitochondrialnych jest jednak kodowana przez genom jądrowy i importowana do organellum po syntezie w cytoplazmie.
Szczególnym aspektem mitochondrialnego DNA jest sposób jego dziedziczenia. U większości gatunków, w tym u człowieka, mtDNA przekazywane jest niemal wyłącznie w linii matczynej. Wynika to z faktu, że plemnik wnosi do zygoty przede wszystkim jądro z materiałem genetycznym, natomiast mitochondria zarodka pochodzą głównie z komórki jajowej. Dzięki temu mtDNA zawiera swoistą „kronikę” rodowodów matczynych, wykorzystywaną w badaniach antropologicznych, genealogicznych i ewolucyjnych.
Mutacje w DNA mitochondrialnym mogą prowadzić do wielu chorób, często dotyczących tkanek o dużym zapotrzebowaniu energetycznym, takich jak mięśnie, serce czy układ nerwowy. Objawy mogą być różnorodne: od osłabienia mięśni, przez zaburzenia neurologiczne, aż po kardiomiopatie. Charakterystyczną cechą jest heteroplazmia, czyli współistnienie w jednej komórce cząsteczek mtDNA prawidłowego i zmutowanego. Proporcja między nimi decyduje o nasileniu objawów.
Choroby mitochondrialne mogą wynikać zarówno z mutacji w mtDNA, jak i z defektów genów jądrowych kodujących białka mitochondrialne. Diagnostyka jest skomplikowana i opiera się na połączeniu badań genetycznych, biochemicznych i obrazowych. Terapie przyczynowe są wciąż ograniczone, choć intensywnie rozwijają się strategie oparte na modyfikacjach genetycznych, przeszczepach mitochondriów lub manipulacji biogenezą tych organelli.
Interesującym przykładem wykorzystania wiedzy o dziedziczeniu mitochondrialnym są techniki wspomaganego rozrodu, znane potocznie jako „dziecko trojga rodziców”. W niektórych przypadkach, aby uniknąć przekazania ciężkich chorób mitochondrialnych, wykorzystuje się komórkę jajową dawczyni z prawidłowymi mitochondriami, łącząc ją z jądrem komórkowym matki. Takie procedury rodzą liczne pytania etyczne i prawne, ale jednocześnie pokazują, jak głęboko rafinowana wiedza o mitochondriach wnika w praktykę medyczną.
Mutacje mitochondrialne i kumulacja uszkodzeń mtDNA są wiązane z procesem starzenia. Jedna z hipotez głosi, że z czasem dochodzi do stopniowego pogarszania się funkcji łańcucha oddechowego, wzrostu produkcji ROS i pogłębiającego się deficytu energetycznego. Wpływa to na działanie tkanek o wysokim metabolizmie, takich jak mięśnie, mózg czy serce. Choć hipsoteza mitochondrialna starzenia jest obecnie modyfikowana i kwestionowana w części aspektów, mitochondria pozostają ważnym elementem układanki wyjaśniającej mechanizmy starzenia organizmów.
Nie bez znaczenia jest także rola mitochondriów w chorobach przewlekłych, takich jak cukrzyca typu 2, choroby neurodegeneracyjne czy otyłość. Dysfunkcja w metabolizmie mitochondrialnym, zaburzenia w biogenezie organelli lub w ich degradacji przez mitofagię mogą wpływać na wrażliwość komórek na insulinę, zdolność neuronów do przetrwania czy regulację masy ciała. Badania nad tymi zależnościami są intensywnie rozwijane, a mitochondria stają się często celem potencjalnych terapii farmakologicznych.
Mitochondria, adaptacja i perspektywy badań
Mitochondria nie są sztywnymi strukturami o raz na zawsze ustalonej liczbie i aktywności. Komórki potrafią dynamicznie regulować ich ilość i jakość w odpowiedzi na bodźce środowiskowe, wysiłek fizyczny, dietę czy stres. Proces powstawania nowych mitochondriów nazywamy biogenezą mitochondrialną. Jest on kontrolowany przez złożoną sieć czynników transkrypcyjnych i koaktywatorów, z których jednym z najlepiej poznanych jest białko PGC-1α. Aktywacja tego szlaku prowadzi do zwiększenia liczby i wydajności mitochondriów.
Przykładem adaptacji mitochondrialnej jest trening wytrzymałościowy. Regularny wysiłek fizyczny zwiększa liczbę mitochondriów w mięśniach, poprawia ich zdolność do utleniania kwasów tłuszczowych i wytwarzania ATP, co przekłada się na lepszą wydolność organizmu. Z drugiej strony, siedzący tryb życia i dieta bogata w nadmiar kalorii, ale uboga w składniki odżywcze, mogą prowadzić do spadku jakości funkcjonowania mitochondriów, zwiększonej produkcji ROS i zaburzeń metabolicznych.
Utrzymanie populacji zdrowych mitochondriów w komórce wymaga równowagi między biogenezą a degradacją uszkodzonych organelli. Proces selektywnego usuwania uszkodzonych mitochondriów nazywa się mitofagią i jest formą autofagii. Dzięki niemu komórka pozbywa się organelli, które nie funkcjonują prawidłowo, zapobiegając nadmiernej produkcji ROS i dalszym uszkodzeniom. Zaburzenia mitofagii wiązane są z wieloma chorobami neurodegeneracyjnymi, w tym z chorobą Parkinsona.
W ostatnich latach coraz więcej uwagi poświęca się roli mitochondriów w układzie odpornościowym. Mitochondrialne DNA, gdy dostanie się do cytoplazmy lub przestrzeni pozakomórkowej, może być rozpoznawane przez receptory wrodzonej odporności jako sygnał zagrożenia, wywołując reakcję zapalną. Z jednej strony pomaga to w obronie przed infekcjami, z drugiej jednak może przyczyniać się do rozwoju chorób autoimmunizacyjnych i przewlekłego stanu zapalnego.
Postęp technik badawczych, takich jak mikroskopia superrozdzielcza, sekwencjonowanie jednocząsteczkowe czy edycja genomu, umożliwia coraz dokładniejsze śledzenie struktury i funkcjonowania mitochondriów w żywych komórkach. Wprowadzenie narzędzi takich jak CRISPR/Cas dla mitochondrialnego DNA jest trudniejsze niż dla genomu jądrowego, ale rozwijają się alternatywne technologie, np. nukleazy specyficzne dla mitochondriów. Pozwalają one selektywnie usuwać zmutowane kopie mtDNA, co może stać się podstawą przyszłych terapii genowych w chorobach mitochondrialnych.
Interesującym kierunkiem badań jest także próba wykorzystania mitochondriów jako celów w terapii przeciwnowotworowej. Komórki nowotworowe często charakteryzują się zmienionym metabolizmem mitochondrialnym, inną równowagą między glikolizą a fosforylacją oksydacyjną oraz odmienną regulacją śmierci komórkowej. Opracowanie leków, które wybiórczo zaburzałyby mitochondria komórek nowotworowych, pozostawiając względnie nienaruszone mitochondria komórek zdrowych, mogłoby znacząco poprawić skuteczność terapii i zmniejszyć działania niepożądane.
Perspektywy badań nad mitochondriami obejmują także zagadnienia związane z długowiecznością i medycyną personalizowaną. Analiza profilu funkcjonalnego mitochondriów u konkretnej osoby – obejmująca wydajność łańcucha oddechowego, poziom produkcji ROS, zdolność do biogenezy i mitofagii, a także sekwencję mtDNA – może w przyszłości pomóc w przewidywaniu ryzyka chorób, doborze optymalnej diety, aktywności fizycznej czy terapii farmakologicznej. Mitochondria stają się więc nie tylko obiektem badań podstawowych, lecz także potencjalnym narzędziem w praktyce klinicznej.
FAQ – najczęstsze pytania o mitochondria
Czym dokładnie jest mitochondrium i dlaczego nazywa się je „elektrownią” komórki?
Mitochondrium to organellum komórkowe otoczone podwójną błoną, występujące w komórkach eukariotycznych. W jego wnętrzu zachodzi szereg reakcji biochemicznych prowadzących do wytwarzania ATP, głównej cząsteczki przenoszącej energię w komórce. Dzięki temu mitochondria zapewniają komórce paliwo do wszystkich procesów życiowych: syntezy białek, skurczu mięśni, przewodzenia impulsów nerwowych czy transportu aktywnego przez błony.
Skąd wiadomo, że mitochondria pochodzą od bakterii?
O endosymbiotycznym pochodzeniu mitochondriów świadczy kilka niezależnych linii dowodów. Mają własne, najczęściej koliste DNA przypominające bakteryjne, rybosomy podobne do prokariotycznych i dzielą się przez podział przypominający podział komórki bakteryjnej. Otacza je podwójna błona, co pasuje do scenariusza wchłonięcia dawnej bakterii przez komórkę gospodarza. Analizy sekwencji genów mitochondrialnych wskazują na ich pokrewieństwo z określonymi grupami bakterii tlenowych.
Dlaczego mitochondria dziedziczymy głównie po matce?
Podczas zapłodnienia zygota otrzymuje mitochondria przede wszystkim z komórki jajowej, która jest dużo większa i zawiera liczne organella. Plemnik dostarcza głównie jądro z materiałem genetycznym, a jego mitochondria zazwyczaj ulegają degradacji po wniknięciu do komórki jajowej. W efekcie mitochondrialne DNA przekazywane jest niemal wyłącznie w linii matczynej. Ta cecha umożliwia śledzenie rodowodów matczynych i badanie migracji populacji ludzkich w skali ewolucyjnej.
Jakie są główne objawy chorób mitochondrialnych?
Choroby mitochondrialne najczęściej dotykają narządów o wysokim zapotrzebowaniu na energię, dlatego objawy obejmują osłabienie mięśni, nietolerancję wysiłku, zaburzenia neurologiczne, problemy ze wzrokiem lub słuchem, a także kardiomiopatie i arytmie. Mogą pojawiać się również zaburzenia wątroby, nerek czy układu hormonalnego. Obraz kliniczny jest bardzo zróżnicowany, ponieważ nasilenie objawów zależy od proporcji między prawidłowym a zmutowanym DNA mitochondrialnym w poszczególnych tkankach.
Czy można poprawić funkcjonowanie mitochondriów stylem życia?
Na kondycję mitochondriów silnie wpływa styl życia. Regularny wysiłek wytrzymałościowy zwiększa liczbę mitochondriów w mięśniach i ich wydolność, co poprawia ogólną sprawność metaboliczną. Zbilansowana dieta bogata w warzywa, owoce, zdrowe tłuszcze i odpowiednią ilość białka dostarcza niezbędnych składników dla pracy łańcucha oddechowego i systemów antyoksydacyjnych. Unikanie przewlekłego stresu, toksyn oraz nadmiernej podaży kalorii może ograniczać stres oksydacyjny i wspierać procesy naprawcze w mitochondriach.

