Elektrofile należą do najważniejszych pojęć w nowoczesnej chemii, odgrywając kluczową rolę w opisie reakcji organicznych i nieorganicznych. Zrozumienie natury cząstek, które poszukują elektronów, pozwala przewidywać przebieg reakcji, projektować nowe leki, opracowywać materiały funkcjonalne i optymalizować procesy przemysłowe. Poniższy tekst zagłębia się w definicje, teorie, przykłady oraz praktyczne zastosowania elektrofilów, łącząc perspektywę chemii fizycznej, organicznej i biochemii.
Podstawowa definicja i istota elektrofilów
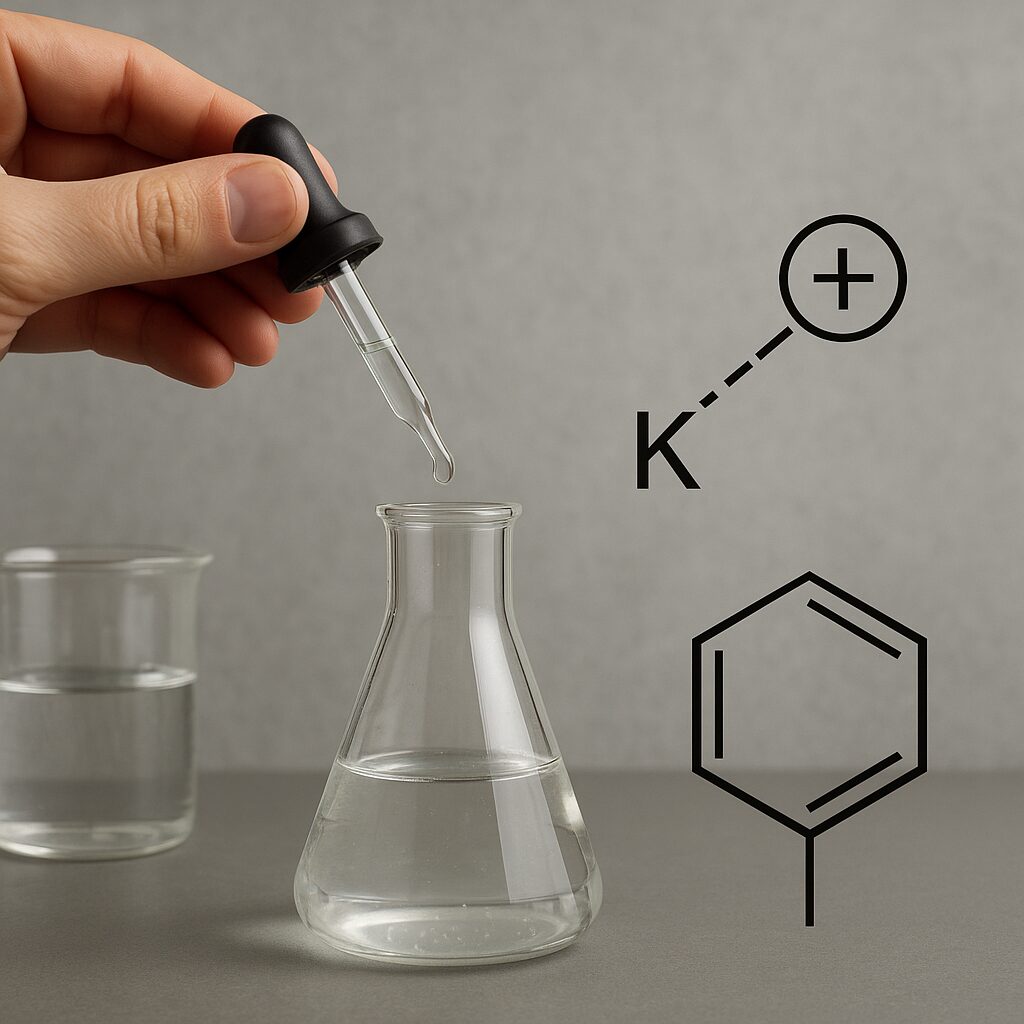
Pojęcie elektrofilu wywodzi się z języka greckiego: „elektron” – ładunek ujemny oraz „philos” – lubiący. Elektrofil to zatem cząstka „lubiąca” elektrony, czyli taka, która dąży do przyjęcia pary elektronowej. W praktyce elektrofilem może być jon dodatni, cząsteczka obojętna z wyraźnym niedoborem elektronów lub fragment większej cząsteczki, w którym zlokalizowany jest silny deficyt ładunku ujemnego.
W ujęciu ogólnym elektrofil to:
- gatunek chemiczny, który akceptuje parę elektronową
- reagent reagujący z nukleofilem, będącym dawcą tej pary
- czynnik inicjujący wiele reakcji, takich jak substytucja, addycja czy sprzęganie
Zgodnie z teorią kwasów i zasad Lewisa, elektrofil jest po prostu kwasem Lewisa – akceptorem pary elektronowej. Taki opis obejmuje zarówno proste jony, jak i bardziej złożone układy molekularne. Dzięki temu chemicy mogą porządkować różne reakcje według wspólnego schematu: nukleofil (baza Lewisa) atakuje elektrofil (kwas Lewisa), prowadząc do powstania nowego wiązania.
Teoretyczne podstawy: elektrofile w świetle różnych ujęć
Teoria kwas–zasada Lewisa
Najbardziej ogólne podejście do elektrofilu opiera się na koncepcji kwasów i zasad Lewisa. W tej teorii:
- kwas Lewisa – akceptor pary elektronowej – odpowiada elektrofilowi
- zasada Lewisa – donor pary elektronowej – odpowiada nukleofilowi
Przykładem może być reakcja pomiędzy jonem H⁺ a cząsteczką amoniaku NH₃. Amoniak posiada wolną parę elektronową na atomie azotu i działa jako nukleofil. Proton H⁺ pozbawiony jest elektronów i stanowi silny elektrofil. Po przyłączeniu protonu do amoniaku powstaje kation amoniowy NH₄⁺. W ujęciu Lewisa obserwujemy przekazanie pary elektronowej od nukleofila do elektrofilu i utworzenie nowego wiązania N–H.
Teoria orbitali molekularnych (HOMO–LUMO)
W nowocześniejszym ujęciu elektofile opisuje się przy użyciu teorii orbitali molekularnych. Kluczowe są tu pojęcia:
- HOMO (Highest Occupied Molecular Orbital) – najwyżej położony obsadzony orbital nukleofila
- LUMO (Lowest Unoccupied Molecular Orbital) – najniżej położony nieobsadzony orbital elektrofilu
Reakcja nukleofil–elektrofil ma miejsce, gdy HOMO nukleofila nachodzi przestrzennie na LUMO elektrofilu i różnica energii między tymi orbitalami jest odpowiednio mała. Im niższa energia LUMO elektrofilu, tym reaktywność elektrofilowa zazwyczaj większa. Ta perspektywa pozwala przewidywać, które fragmenty cząsteczki będą najbardziej podatne na atak nukleofilowy, a które będą wykazywać charakter elektrofilowy.
Teoria HSAB (twarde i miękkie kwasy i zasady)
Teoria HSAB (Hard and Soft Acids and Bases) stanowi kolejne narzędzie do klasyfikowania elektrofilów. W tym podejściu:
- „twarde” elektrofile – małe, silnie spolaryzowane, o wysokim ładunku dodatnim
- „miękkie” elektrofile – większe, bardziej polaryzowalne, często o rozłożonym ładunku
Przykładami twardych elektrofilów są H⁺, Al³⁺, BF₃, podczas gdy miękkimi elektrofilami są np. kationy metali przejściowych o niskim stopniu utlenienia (Pd²⁺ w złożonych ligandach) czy częściowo dodatnio naładowane atomy w związkach organicznych, takie jak węgiel karbonylowy w reaktywnych aldehydach aromatycznych. Zasada HSAB mówi, że twarde nukleofile preferują twarde elektrofile, a miękkie – miękkie, co ułatwia przewidywanie selektywności reakcji.
Rodzaje i klasyfikacja elektrofilów
Elektrofile kationowe
Najprostszą i najbardziej intuicyjną grupą są elektrofile kationowe, czyli jony dodatnie. Charakteryzują się one wyraźnym niedoborem elektronów, co czyni je szczególnie skłonnymi do przyjmowania par elektronowych od nukleofila.
- Proton H⁺ – jeden z najsilniejszych elektrofilów; uczestniczy w niezliczonej liczbie reakcji kwasowo–zasadowych i protonacyjnych
- Kationy metali – np. Fe³⁺, Al³⁺, Zn²⁺; pełnią rolę centrów elektrofilowych w kompleksach, katalizie i reakcjach koordynacyjnych
- Karbenium i karbokationy – jony organiczne o dodatnim ładunku na atomie węgla, niezwykle aktywne w reakcjach substytucji i przekształceń szkieletu węglowego
Takie elektrofile odgrywają ważną rolę w klasycznych reakcjach organicznych, jak SN1 czy E1, gdzie stabilność karbokationu determinuje szybkość i kierunek procesu.
Elektrofile obojętne
Nie każdy elektrofil musi mieć formalny ładunek dodatni. Często cząstki obojętne wykazują silny charakter elektrofilowy z powodu polaryzacji wiązań lub obecności wolnych orbitali. Typowe przykłady to:
- Cząsteczki karbonylowe – takie jak aldehydy i ketony; atom węgla w grupie C=O jest ubogi w elektrony z powodu przesunięcia gęstości elektronowej w stronę bardziej elektroujemnego tlenu
- Halogenki acylowe – R–CO–Cl; połączenie grupy karbonylowej z dobrym odchodzącym nukleofilem (Cl⁻) czyni węgiel karbonylowy wyjątkowo podatnym na atak
- Trójskoordynowane związki boru – np. BF₃, BCl₃, BR₃; atom boru ma nieskompletowaną oktetową powłokę elektronową, co czyni go silnym akceptorem elektronów
W wielu takich przypadkach elektrofilowość jest konsekwencją różnicy elektroujemności pomiędzy atomami, co powoduje powstanie dipolu i częściowego ładunku dodatniego na jednym z nich.
Elektrofilowe fragmenty związków organicznych
W złożonych cząsteczkach organicznych często tylko wybrane fragmenty zachowują się jak elektrofile. Typowe motywy to:
- atom węgla grupy karbonylowej C=O
- atom węgla w wiązaniu C–X, gdzie X jest dobrym odchodzącym nukleofilem (Cl, Br, I, OTs)
- atom węgla w związkach z grupą nitrową, sulfonową czy estrową
- częściowo dodatnio naładowane atomy węgla przy pierścieniach aromatycznych, aktywowanych czynnikami odciągającymi elektrony
Rozpoznanie tych centrów elektrofilowych jest podstawowym narzędziem projektowania syntez organicznych i przewidywania miejsc ataku nukleofila, co jest niezwykle przydatne w planowaniu ścieżek do nowych substancji, w tym leków i materiałów polimerowych.
Mechanizmy reakcji elektrofilowych
Substytucja elektrofilowa w chemii aromatycznej
Jednym z kluczowych mechanizmów, w których elektrofile odgrywają centralną rolę, jest substytucja elektrofilowa aromatyczna (SEAr). Przykłady obejmują nitrowanie, sulfonowanie, halogenowanie i alkilowanie pierścieni benzenowych.
Ogólny przebieg reakcji można przedstawić następująco:
- wytworzenie silnego elektrofilu (np. NO₂⁺ w nitrowaniu)
- atak elektrofilu na bogaty w elektrony pierścień aromatyczny
- powstanie kompleksu σ (karbokationu aromatycznego)
- deprotonacja i odtworzenie aromatyczności
W klasycznym nitrowaniu benzenu mieszaniną stężonego kwasu azotowego i siarkowego powstaje jon nitroniowy NO₂⁺, będący wyjątkowo silnym elektrofilem. Atakuje on pierścień benzenowy, tworząc niestabilny kompleks σ, który następnie traci proton, przywracając aromatyczność i tworząc nitrobenzen. Obecność podstawników na pierścieniu (np. CH₃, OH, NO₂) wpływa na gęstość elektronową i selektywność ataku elektrofilu, co pozwala precyzyjnie kierować reakcję w stronę orto, meta lub para.
Substytucja elektrofilowa alifatyczna
Choć rzadziej omawiana niż substytucja nukleofilowa, substytucja elektrofilowa alifatyczna również istnieje i jest istotna w specyficznych układach, np. w związkach zawierających silne grupy odciągające elektrony. W takich przypadkach atom węgla łańcucha alifatycznego może uzyskać charakter elektrofilowy i zostać zaatakowany przez elektronoofobowe cząsteczki, często w warunkach katalizy kwasowej lub w obecności utleniaczy.
Mechanizmy tych reakcji bywają złożone i obejmują przegrupowania, tworzenie krótkotrwałych karbokationów lub udział wolnych rodników. Mimo mniejszej częstości występowania niż klasyczne SN1 i SN2, zrozumienie elektrofilowej natury centrów reaktywnych w alifatycznych układach pozwala rozszerzyć możliwości syntezy złożonych struktur organicznych.
Addycja elektrofilowa do wiązań wielokrotnych
Kolejnym niezwykle ważnym mechanizmem jest addycja elektrofilowa do wiązań C=C i C≡C. W alkenach i alkinach obecność wiązania π, bogatego w elektrony, sprzyja ich atakowi przez elektrofile.
Ogólny schemat addycji elektrofilowej do alkenu wygląda następująco:
- elektrofil atakuje wiązanie π, tworząc karbokation
- nukleofil atakuje powstały karbokation, dając produkt addycji
Przykładem jest addycja HBr do prostej cząsteczki, takiej jak eten. Proton H⁺ pełni rolę elektrofilu i przyłącza się do jednego z atomów węgla, generując karbokation na drugim. Następnie jon bromkowy Br⁻ działa jako nukleofil, uzupełniając drugi etap reakcji. W bardziej złożonych układach reguła Markownikowa opisuje, na który atom węgla trafi proton, a na który reszta nukleofilowa, co powiązane jest ze stabilnością przejściowego karbokationu.
Reakcje z udziałem elektrofilów w chemii nieorganicznej
Elektrofile odgrywają również istotną rolę w chemii nieorganicznej, szczególnie w reakacjach kompleksowania, utleniania i przegrupowań ligando–metalicznych. Kationy metali przejściowych, takie jak Pt²⁺, Pd²⁺ czy Fe³⁺, często pełnią podwójną rolę: są centrami elektrofilowymi oraz katalizatorami. Wiązanie ligandów do tych centrów można traktować jako klasyczną reakcję elektrofil–nukleofil, w której ligand (np. NH₃, Cl⁻, fosfiny) donuje parę elektronową, a metal ją akceptuje.
W utlenianiu związków organicznych metale w wysokich stopniach utlenienia (MnO₄⁻, Cr₂O₇²⁻) mogą działać jak silne elektrofile, atakując centra bogate w elektrony, np. wiązania podwójne czy grupy alkoholowe. Zrozumienie ich elektrofilowego charakteru umożliwia kontrolowanie selektywności utleniania – np. wybór warunków utleniających pierwotny alkohol do aldehydu albo do kwasu karboksylowego.
Siła elektrofilów: czynniki wpływające i przykłady
Wpływ struktury i elektroujemności
Siła elektrofilowa zależy od zdolności cząstki do przyjmowania elektronów, co jest funkcją kilku czynników:
- elektroujemności atomów w otoczeniu centrum reaktywnego
- obecności grup odciągających elektrony (–NO₂, –CF₃, –CN)
- ładunku formalnego (kationy są zazwyczaj silniejszymi elektrofilami niż cząstki obojętne)
- stabilności powstającego po reakcji kompleksu lub produktu
W grupie związków karbonylowych wprowadzenie silnie elektroujemnych podstawników w sąsiedztwie atomu węgla (np. fluorowców, grupy nitrowej) zwiększa jego elektrofilowość, ułatwiając atak nukleofilowy. Odwrotnie, grupy donorowe, takie jak –OCH₃ czy –NR₂, zmniejszają deficyt elektronowy, osłabiając charakter elektrofilowy centrum.
Środowisko reakcji: rozpuszczalnik i temperatura
Rozpuszczalnik ma istotny wpływ na efektywną elektrofilowość. Rozpuszczalniki polarne protowe (np. woda, alkohole) stabilizują ładunki, co może ułatwiać powstawanie kationowych elektrofilów, ale jednocześnie mogą też kompleksować z nimi, częściowo osłabiając ich reaktywność. Rozpuszczalniki aprotowe polarne (DMF, DMSO, acetonitryl) często sprzyjają reakcjom, w których wymagane jest zachowanie wysokiej aktywności elektrofilów i nukleofilów.
Temperatura wpływa na szybkość reakcji i rozkład energii, ale może również przesuwać równowagi pomiędzy formami bardziej i mniej zjonizowanymi. Wysoka temperatura często sprzyja tworzeniu krótkotrwałych, wysoko reaktywnych cationów, które wykazują wyjątkowo silną elektrofilowość, co jest wykorzystywane w wielu procesach przemysłowych i w syntezie związków aromatycznych.
Przykłady silnych i słabszych elektrofilów
- Silne elektrofile: NO₂⁺, H⁺ w superkwasach, karbokationy tert-butylowe, halogenki acylowe (RCOCl), BF₃, AlCl₃
- Umiarkowane elektrofile: proste aldehydy i ketony, estrów kwasów karboksylowych, niektóre kationy metali przejściowych
- Słabe elektrofile: amidy, karboksylany, słabo spolaryzowane wiązania C–X w związkach nasyconych bez silnych grup odciągających elektrony
Klasyfikacja ta ma charakter orientacyjny i silnie zależy od warunków reakcji, zwłaszcza od obecności katalizatorów, rozpuszczalnika i dodatków, które mogą aktywować lub dezaktywować dane centrum elektrofilowe.
Elektrofile w syntezie organicznej i przemyśle
Projektowanie leków i interakcje z biomolekułami
W chemii medycznej elektrofile odgrywają szczególną rolę w projektowaniu leków działających na zasadzie kowalencyjnej modyfikacji białek. Cząsteczki zawierające reaktywne centra elektrofilowe mogą tworzyć trwałe wiązania z nukleofilowymi resztami aminokwasów (np. cysteina, seryna, lizyna) w centrach aktywnych enzymów. Pozwala to uzyskać silne i długotrwałe zahamowanie działania danego białka.
Przykładami są:
- inhibitory proteaz z grupami epoksydowymi lub halogenoketonowymi
- inhibitory kinaz zawierające akrylany lub inne układy akceptorowe dla nukleofilowych grup tiolowych
- leki przeciwnowotworowe oparte na związkach platyny, gdzie centrum metaliczne działa jako silny elektrofil
Projektując tego typu cząsteczki, chemicy muszą starannie wyważyć elektrofilowość, aby reakcja była wystarczająco selektywna i ograniczała niepożądane modyfikacje innych białek w organizmie. Zbyt silny elektrofil może prowadzić do toksyczności i efektów ubocznych, dlatego precyzyjne modelowanie oddziaływań nukleofil–elektrofil jest kluczowe.
Elektrofile w syntezie materiałów i polimerów
W produkcji polimerów elektrofile uczestniczą w inicjacji i propagacji łańcuchów. Przykładowo, w kationowej polimeryzacji alkenów (np. styrenu, izobutylenu) pierwszym etapem jest wytworzenie kationowego centrum elektrofilowego, które następnie atakuje kolejne monomery, prowadząc do wzrostu łańcucha. Stabilność tego kationu i jego reaktywność wobec wiązań C=C monomerów przesądzają o strukturze, masie molowej i właściwościach powstającego tworzywa.
Elektrofile są też kluczowe w syntezie materiałów o kontrolowanych właściwościach elektronicznych, takich jak przewodzące polimery czy materiały do elektroniki organicznej. Wprowadzanie silnie elektrofilowych fragmentów do łańcuchów polimerowych wpływa na energię poziomów HOMO i LUMO, a tym samym na przewodnictwo, barwę, stabilność i zdolność transportu ładunku w materiałach.
Procesy przemysłowe i kataliza
W skali przemysłowej elektrofile uczestniczą w wielu kluczowych procesach, takich jak:
- alkilowanie i acylowanie aromatów (np. reakcje Friedla–Craftsa)
- produkcja barwników i pigmentów poprzez elektrofilową modyfikację pierścieni aromatycznych
- synteza środków powierzchniowo czynnych, plastyfikatorów, środków ochrony roślin
Katalizatory kwasowo–Lewisowskie, takie jak AlCl₃, FeCl₃ czy kompleksy metali przejściowych, zwiększają elektrofilowość substratów przez tworzenie kompleksów aktywowanych. Przykładowo, w reakcji acylowania Friedla–Craftsa kompleks RCOCl z AlCl₃ generuje silny kation acyliowy, który następnie reaguje z pierścieniem aromatycznym. Reguły selektywności elektrofilowych reakcji aromatycznych są wykorzystywane do produkcji konkretnych izomerów z wysoką wydajnością.
Rola elektrofilów w chemii środowiska i biochemii
Elektrofile jako czynniki toksyczne i mutagenne
W chemii środowiskowej liczne związki zanieczyszczające powietrze, wodę i glebę wykazują elektrofilowy charakter. Reaktywne aldehydy, epoksydy, halogenowane węglowodory czy metabolity wielopierścieniowych węglowodorów aromatycznych mogą reagować z nukleofilowymi fragmentami DNA i białek, prowadząc do mutacji, uszkodzeń komórek i rozwoju nowotworów.
Metabolizm ksenobiotyków w organizmie często polega na przekształcaniu ich w bardziej hydrofilne formy, ale niektóre pośrednie produkty utleniania stają się silnie elektrofilowe, co zwiększa ich zdolność do tworzenia kowalencyjnych adduktów z biomolekułami. Analiza tych interakcji jest podstawą toksykologii i oceny ryzyka chemicznego w środowisku.
Elektrofilowe centra w enzymach i metabolitach
W samej biochemii liczne metabolity i kofaktory zawierają fragmenty o charakterze elektrofilowym. Przykłady obejmują:
- aldehydowe formy cukrów (np. w glikolizie)
- aktywowane estry kwasów karboksylowych, takie jak acyloaddukty koenzymu A
- substraty aktywowane przez ATP, w których powstają reaktywne fosfoanhydrydy
Enzymy często stabilizują w swoich centrach aktywnych przejściowe elektrofilowe formy substratów, kierując przepływem elektronów i protonów w stronę wysoce specyficznych produktów. Z punktu widzenia projektowania inhibitorów enzaymów, identyfikacja tych chwilowych elektrofilowych centrów pozwala opracować leki naśladujące stan przejściowy reakcji.
Jak rozpoznawać elektrofile w praktyce laboratoryjnej
Analiza strukturalna i spektroskopowa
W praktyce laboratoryjnej chemicy posługują się kombinacją wiedzy teoretycznej oraz danych eksperymentalnych, by określić, które fragmenty cząsteczki zachowują się jak elektrofile. Podstawowe narzędzia to:
- analiza struktury z wykorzystaniem reguł elektronowych (oktet, rezonans, indukcja)
- spektroskopia NMR – zmiany przesunięć chemicznych wskazujące na deficyt elektronów przy określonych atomach
- spektroskopia IR – położenie pasm C=O, C=N i innych, zależne od elektronowego otoczenia
- obliczenia kwantowo–chemiczne – wyznaczenie energii LUMO i map gęstości ładunku
Połączenie tych metod pozwala utożsamić centra, w których istnieje najwyższe prawdopodobieństwo ataku nukleofila, a tym samym rozpoznać elementy elektrofilowe. W syntezie złożonych cząsteczek, takich jak naturalne produkty roślinne czy makrocykliczne leki, takie analizy są niezbędne.
Testy reaktywności i modele modelowe
Drugim podejściem jest eksperymentalne testowanie reaktywności. Stosując proste nukleofile modelowe (np. aniony tiolanowe, aminy, alkohole), bada się, które fragmenty analizowanej cząsteczki wchodzą w reakcję addycji lub substytucji. Obserwacja szybkości i selektywności tych reakcji, wsparta analizą produktów, pozwala zweryfikować hipotezy dotyczące elektrofilowego charakteru centrum.
Dodatkowo korelacje liniowe wolnej energii (np. równania Hammetta, Tafta) umożliwiają ilościowe powiązanie wpływu podstawników z elektrofilowością centrum reaktywnego. W ten sposób można systematycznie modyfikować strukturę związków w celu optymalizacji ich zachowania, czy to w syntezie, czy w zastosowaniach biologicznych.
FAQ – najczęściej zadawane pytania o elektrofile
Czym różni się elektrofil od nukleofila?
Elektrofil to gatunek chemiczny poszukujący elektronów, czyli akceptor pary elektronowej; w teorii Lewisa jest to kwas. Nukleofil jest jego przeciwieństwem – jest donorem pary elektronowej, czyli zasadą Lewisa. W reakcji tworzenia nowego wiązania nukleofil atakuje elektrofil, przesuwając parę elektronową w kierunku cząstki ubogiej w elektrony. Ta relacja jest podstawą opisu większości reakcji organicznych i nieorganicznych.
Jak rozpoznać, że dana cząsteczka jest elektrofilem?
Elektrofil często posiada formalny ładunek dodatni lub częściowy deficyt elektronów na konkretnym atomie, np. węgla karbonylowego. O obecności centrum elektrofilowego świadczą silnie spolaryzowane wiązania, obecność grup odciągających elektrony oraz niski poziom energetyczny orbitali LUMO. W praktyce korzysta się z analizy struktury, danych spektroskopowych i testów reaktywności z prostymi nukleofilami, aby zidentyfikować najbardziej podatne miejsca ataku.
Dlaczego elektrofile są tak ważne w syntezie organicznej?
Reakcje między nukleofilami a elektrofilami pozwalają tworzyć nowe wiązania C–C, C–N, C–O i wiele innych, co stanowi fundament syntezy związków organicznych. Kontrolując strukturę i siłę elektrofilów, chemicy mogą decydować, w którym miejscu cząsteczki dojdzie do reakcji oraz jakie grupy funkcyjne zostaną wprowadzone. To z kolei umożliwia planowanie wieloetapowych ścieżek syntezy leków, barwników, materiałów polimerowych i związków o złożonej architekturze molekularnej.
Czy elektrofile zawsze są groźne dla organizmu?
Elektrofile mogą być zarówno korzystne, jak i szkodliwe. W organizmach wiele metabolitów i kofaktorów zawiera kontrolowane centra elektrofilowe, niezbędne w reakcjach enzymatycznych. Jednak silnie reaktywne elektrofile, zwłaszcza powstające podczas metabolizmu ksenobiotyków, mogą atakować DNA i białka, powodując uszkodzenia komórkowe. Dlatego w projektowaniu leków i ocenie toksyczności związków duży nacisk kładzie się na zrozumienie ich elektrofilowego charakteru oraz selektywności reakcji.
Jak można regulować siłę elektrofilów w praktyce chemicznej?
Siłę elektrofilów można modyfikować poprzez dobór odpowiednich podstawników, zmianę warunków reakcji i zastosowanie katalizatorów. Wprowadzanie grup odciągających elektrony zwiększa elektrofilowość, natomiast grupy donorowe ją obniżają. Użycie kwasów Lewisa lub Brønsteda pozwala aktywować określone centra, np. karbonylowe, tworząc bardziej reaktywne kompleksy. Z kolei dobór rozpuszczalnika, temperatury i stężenia reagentów umożliwia precyzyjne dostrojenie szybkości i selektywności reakcji elektrofilowych.