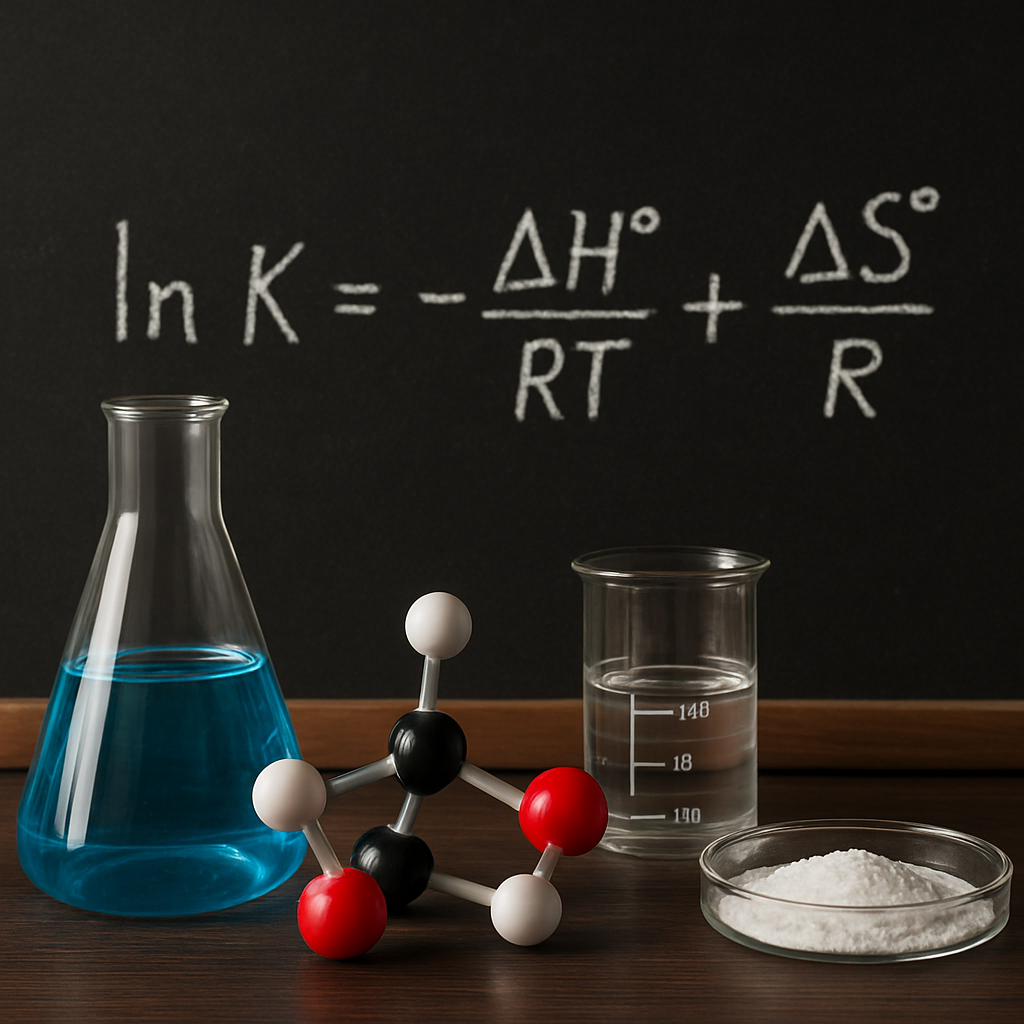
Równanie van ’t Hoffa należy do najważniejszych zależności w termodynamice chemicznej. Pozwala powiązać stałą równowagi reakcji z temperaturą i tym samym przewidzieć, jak zmiana warunków termicznych wpłynie na przesunięcie równowagi chemicznej. Dzięki temu stanowi pomost między opisem doświadczalnym a głębokim rozumieniem mechanizmów reakcji, ich wydajności, a nawet stabilności materiałów czy biomolekuł w różnych temperaturach.
Historyczne i teoretyczne podstawy równania van ’t Hoffa
Twórcą równania jest Jacobus Henricus van ’t Hoff, holenderski chemik i pierwszy laureat Nagrody Nobla z chemii (1901). Zajmował się on zależnością pomiędzy ciśnieniem osmotycznym a temperaturą, a także wpływem temperatury na stałe równowagi reakcji. Analiza tych zależności doprowadziła do sformułowania wzoru, który z czasem stał się podstawowym narzędziem w termodynamice chemicznej i inżynierii procesowej.
Podstawą równania van ’t Hoffa jest powiązanie pojęcia stałej równowagi K z funkcjami termodynamicznymi, szczególnie z energią swobodną Gibbsa, oznaczaną ΔG. Dla reakcji w warunkach standardowych zachodzi związek:
ΔG° = −RT ln K
gdzie R to uniwersalna stała gazowa, T to temperatura w kelwinach, a K to stała równowagi. Z kolei zmiana energii swobodnej związana jest z entalpią i entropią:
ΔG° = ΔH° − TΔS°
Po połączeniu tych dwóch równań i uporządkowaniu otrzymujemy zależność, która po zróżniczkowaniu względem temperatury prowadzi do klasycznej postaci równania van ’t Hoffa:
d(ln K)/dT = ΔH° / (RT²)
Zakłada się tu, że standardowa entalpia reakcji ΔH° jest w określonym przedziale temperatur w przybliżeniu stała. To jedno z kluczowych założeń, które ogranicza, ale jednocześnie bardzo upraszcza praktyczne zastosowanie równania.
Równanie van ’t Hoffa – wyprowadzenie i interpretacja termodynamiczna
Wyprowadzenie różniczkowe
Punktem wyjścia jest połączenie relacji na energię swobodną i stałą równowagi:
−RT ln K = ΔH° − TΔS°
Po przekształceniu otrzymuje się:
ln K = −ΔH°/(RT) + ΔS°/R
Jest to forma przypominająca równanie prostej (y = ax + b), gdzie:
- y ≡ ln K,
- x ≡ 1/T,
- a ≡ −ΔH°/R,
- b ≡ ΔS°/R.
Jeżeli zróżniczkujemy postać ln K względem temperatury, przy założeniu stałości ΔH° i ΔS°, otrzymujemy:
d(ln K)/dT = ΔH° / (RT²)
To właśnie klasyczne różniczkowe równanie van ’t Hoffa. Pokazuje ono jednoznacznie, że szybkość zmian ln K z temperaturą jest wprost proporcjonalna do entalpii reakcji i odwrotnie proporcjonalna do kwadratu temperatury.
Postać zintegrowana i wykres van ’t Hoffa
W praktyce częściej używa się zintegrowanej postaci równania, która pozwala porównywać wartości stałej równowagi w dwóch temperaturach T₁ i T₂. Całkując wyrażenie d(ln K)/dT = ΔH°/(RT²), otrzymujemy:
ln(K₂/K₁) = −ΔH°/R (1/T₂ − 1/T₁)
Ta postać umożliwia obliczenie nieznanej stałej równowagi K₂ w temperaturze T₂, jeśli znamy K₁ w temperaturze T₁ i wartość ΔH°. Jest to szczególnie cenne dla laboratoriów i przemysłu, gdzie pomiar K w wielu temperaturach może być kłopotliwy lub kosztowny.
Przekształcając równanie ln K = −ΔH°/(RT) + ΔS°/R, można sporządzić tzw. wykres van ’t Hoffa, polegający na naniesieniu ln K w funkcji 1/T. Otrzymana prosta ma nachylenie równe −ΔH°/R i punkt przecięcia z osią ln K równy ΔS°/R. Z takiej prostej można więc wyznaczyć entalpię i entropię reakcji na podstawie zależności stałej równowagi od temperatury.
Interpretacja fizyczna – znak entalpii i kierunek przesunięcia równowagi
Znak ΔH° ma kluczowe znaczenie dla interpretacji równania van ’t Hoffa:
- Dla reakcji egzotermicznej (ΔH° < 0) wzrost temperatury zmniejsza wartość K – równowaga przesuwa się w stronę substratów.
- Dla reakcji endotermicznej (ΔH° > 0) wzrost temperatury zwiększa wartość K – równowaga przesuwa się w stronę produktów.
Zachowanie to jest zgodne z zasadą Le Chateliera-Brauna: układ przeciwdziała wprowadzonym zmianom. Dostarczenie ciepła sprzyja więc reakcjom endotermicznym, które to ciepło pochłaniają, natomiast podwyższona temperatura ogranicza przebieg reakcji egzotermicznych, które ciepło wydzielają.
Zastosowania i ograniczenia równania van ’t Hoffa
Od kinetyki do termodynamiki – związek z równaniem Arrheniusa
Choć równanie van ’t Hoffa jest równaniem termodynamicznym, istnieje ciekawy związek pomiędzy nim a opisem kinetycznym reakcji. Dla prostych reakcji elementarnych stała równowagi jest powiązana ze stosunkiem stałych szybkości reakcji w przód (k₁) i wstecz (k₋₁):
K = k₁/k₋₁
Każda z tych stałych szybkości często spełnia równanie Arrheniusa:
k = A exp(−Eₐ/RT)
gdzie Eₐ to energia aktywacji, a A to tzw. czynnik przedeksponencjalny. Gdy połączymy oba opisy, okaże się, że zmiana K z temperaturą wynika z różnicy energii aktywacji dla reakcji w przód i wstecz. Termodynamiczna entalpia reakcji ΔH° jest zatem w pewnym sensie powiązana z bilansem barier energetycznych w obu kierunkach reakcji. To pokazuje, że równanie van ’t Hoffa stanowi pomost między opisem energetycznym równowagi a opisem kinetycznym szybkości reakcji.
Projektowanie procesów przemysłowych
W chemii przemysłowej równanie van ’t Hoffa jest niezwykle przydatne przy doborze optymalnej temperatury prowadzenia reakcji. Wiele procesów, takich jak synteza amoniaku (proces Habera-Boscha), produkcja kwasu siarkowego czy reforming parowy metanu, opiera się na reakcjach, dla których zarówno równowaga, jak i kinetyka są wrażliwe na temperaturę.
Dla reakcji egzotermicznych wzrost temperatury poprawia zwykle szybkość reakcji, ale jednocześnie obniża wartość stałej równowagi, a więc maksymalną możliwą konwersję substratów. Inżynier procesowy musi znaleźć kompromis: temperaturę, przy której uzyskuje się wystarczającą szybkość przy akceptowalnej wydajności równowagowej. W tym zadaniu z pomocą przychodzi właśnie równanie van ’t Hoffa, które pozwala przewidzieć, jak zmiana temperatury wpłynie na położenie równowagi.
W praktyce przemysłowej korzysta się z równania w formie:
ln(K₂/K₁) = −ΔH°/R (1/T₂ − 1/T₁)
Znając parametry termodynamiczne z danych literaturowych lub wcześniejszych pomiarów, można wyznaczyć wartość K dla serii temperatur i ocenić, czy dana reakcja jest opłacalna przy konkretnych warunkach pracy instalacji.
Biochemia i stabilność białek
Równanie van ’t Hoffa znalazło szerokie zastosowanie w badaniach biochemicznych, zwłaszcza przy analizie równowag wiązania ligand–białko, asocjacji kompleksów makromolekularnych czy procesu fałdowania białek. W takich przypadkach bada się np. zależność stałej dysocjacji Kd (odwrotność stałej wiązania) od temperatury. Po przekształceniu równania można wówczas wyznaczyć entalpię i entropię wiązania.
W eksperymentach typu ITC (kalorymetria izotermiczna) czy DSC (spektroskopia różnicowej kalorymetrii skaningowej) często konfrontuje się dane bezpośrednio pomiarowe z analizą van ’t Hoffa. Jeżeli ΔH° wyznaczona z równania van ’t Hoffa zgadza się z entalpią zmierzoną kalorymetrycznie, sugeruje to prosty model wiązania (np. jedna cząsteczka ligandu na jedno miejsce wiążące w białku). Różnice mogą wskazywać na złożony mechanizm, kooperatywność czy zmiany konformacyjne biomolekuł.
Rozpuszczalność substancji i procesy krystalizacji
W wielu procesach technologicznych kluczowe znaczenie ma rozpuszczalność substancji, która jest wprost związana ze stałą równowagi procesu rozpuszczania. Równanie van ’t Hoffa może służyć do opisu zależności rozpuszczalności od temperatury, zwłaszcza jeśli dysponujemy wartościami entalpii rozpuszczania.
Na przykład dla prostego procesu:
ciało stałe ⇌ jon(y) w roztworze
zmiana temperatury wpłynie na równowagę i tym samym na maksymalne stężenie rozpuszczonej substancji. W praktyce ma to znaczenie dla projektowania operacji krystalizacji, oczyszczania substancji (np. leków), produkcji soli i nawozów, czy też w przemyśle spożywczym, gdzie kontrola rozpuszczania i krystalizacji (np. cukrów) jest kluczowa dla jakości produktu.
Ograniczenia i założenia równania van ’t Hoffa
Mimo ogromnej użyteczności, równanie van ’t Hoffa opiera się na szeregu istotnych założeń, które ograniczają jego stosowalność:
- Stałość entalpii w badanym przedziale temperatur – przyjmuje się, że ΔH° nie zależy od temperatury. W rzeczywistości entalpia może się zmieniać, zwłaszcza przy szerokim zakresie temperatur.
- Idealność roztworów i gazów – w wielu zastosowaniach zakłada się, że roztwory zachowują się idealnie, a aktywności można zastąpić stężeniami lub ciśnieniami. W roztworach rzeczywistych konieczne może być uwzględnienie współczynników aktywności.
- Brak zmian fazowych – równanie stosuje się w zakresie, w którym faza układu nie ulega zmianie (np. brak przejść stałe–ciecz, ciekłe–gaz).
- Brak istotnych zmian mechanizmu reakcji – w zbyt wysokich lub niskich temperaturach mechanizm reakcji może się zmienić, co wpływa na interpretację parametrów ΔH° i ΔS°.
W sytuacjach, gdy zmiana temperatury jest znaczna, należy rozszerzyć opis o zależność entalpii od temperatury, co wymaga uwzględnienia różnicy pojemności cieplnych reagentów i produktów. Prowadzi to do bardziej złożonych równań, ale podstawową ideą nadal pozostaje powiązanie logarytmu stałej równowagi z odwrotnością temperatury.
Równanie van ’t Hoffa a praktyka laboratoryjna
W warunkach laboratoryjnych równanie van ’t Hoffa stanowi ważne narzędzie analizy danych doświadczalnych. Typowym zadaniem jest wyznaczenie entalpii reakcji z pomiarów równowagi w kilku temperaturach. Procedura może wyglądać następująco:
- Wyznaczenie wartości stałej równowagi K w co najmniej trzech różnych temperaturach.
- Obliczenie ln K dla każdej temperatury i wyznaczenie odpowiadających wartości 1/T.
- Sporządzenie wykresu ln K vs. 1/T.
- Wyznaczenie prostej najlepszego dopasowania (metoda regresji liniowej).
- Obliczenie nachylenia prostej m, a następnie ΔH° = −mR.
- Odcięcie od osi ln K daje ΔS°/R, stąd ΔS° = R · b.
Taki sposób analizy jest standardem w dydaktyce chemii fizycznej, ponieważ pozwala studentom połączyć pomiary eksperymentalne z interpretacją termodynamiczną. Jest również wykorzystywany w pracy badawczej, zwłaszcza tam, gdzie trudno jest bezpośrednio zmierzyć entalpię procesów.
Znaczenie równania van ’t Hoffa w szerszym kontekście naukowym
Równanie van ’t Hoffa jest wykorzystywane nie tylko w klasycznej chemii, ale również w takich dziedzinach jak geochemia, nauki o materiałach, farmacja czy nauki środowiskowe. W geochemii pozwala analizować równowagi rozpuszczania minerałów i tworzenia się nowych faz w skorupie ziemskiej. W farmacji pomaga oceniać stabilność substancji czynnych w różnych temperaturach przechowywania, a także badać wiązanie lek–receptor.
W naukach o materiałach równanie to może być stosowane do opisu przemian fazowych, zwłaszcza tam, gdzie istotne są równowagi między różnymi strukturami krystalicznymi czy stopami. Pozwala też oceniać wpływ temperatury na zachowanie materiałów funkcjonalnych, np. membran, sorbentów lub katalizatorów, dla których równowagi adsorpcji i desorpcji są kluczowe dla działania.
FAQ
Jak brzmi podstawowa forma równania van ’t Hoffa?
Podstawowa różniczkowa forma równania van ’t Hoffa to d(ln K)/dT = ΔH°/(RT²), gdzie K jest stałą równowagi, T temperaturą, R uniwersalną stałą gazową, a ΔH° standardową entalpią reakcji. Zależność ta pokazuje, jak logarytm stałej równowagi zmienia się wraz z temperaturą i pozwala przewidzieć, w którą stronę przesunie się równowaga po ogrzaniu lub ochłodzeniu układu.
Do czego służy wykres van ’t Hoffa?
Wykres van ’t Hoffa to przedstawienie zależności ln K od 1/T w postaci liniowej. Nachylenie tej prostej jest równe −ΔH°/R, a punkt przecięcia z osią ln K odpowiada ΔS°/R. Analiza takiego wykresu umożliwia wyznaczenie entalpii i entropii reakcji bez bezpośrednich pomiarów kalorymetrycznych. Jest to szeroko stosowane w chemii fizycznej, biochemii (badanie wiązania ligand–białko) oraz przy projektowaniu procesów przemysłowych.
Jakie są główne ograniczenia równania van ’t Hoffa?
Równanie van ’t Hoffa zakłada m.in. stałość entalpii reakcji w rozważanym zakresie temperatur, idealne zachowanie roztworów lub gazów oraz brak zmian fazowych i mechanizmu reakcji. Gdy zmiany temperatury są duże lub układ jest silnie nieidealny, wyniki mogą być obarczone znacznym błędem. W takich sytuacjach trzeba uwzględniać zmienność pojemności cieplnych i współczynników aktywności, korzystając z bardziej złożonych modeli termodynamicznych.

