Energia swobodna jest jednym z kluczowych pojęć termodynamiki i chemii fizycznej, łączącym w sobie ideę energii, porządku oraz kierunku samorzutnych przemian. Pozwala zrozumieć, dlaczego jedne reakcje chemiczne przebiegają spontanicznie, inne wymagają stałego dostarczania energii, a jeszcze inne w ogóle nie zachodzą w danych warunkach. Dzięki niej chemicy projektują procesy przemysłowe, inżynierowie materiałowi dobierają warunki syntezy, a biochemicy badają mechanizmy podtrzymujące życie na poziomie molekularnym.
Intuicyjne wprowadzenie do pojęcia energii swobodnej
W klasycznej termodynamice opis układów opiera się na kilku podstawowych wielkościach: energii wewnętrznej, entalpii, entropii, temperaturze oraz ciśnieniu. Energia swobodna pojawia się jako kombinacja tych wielkości, dopasowana do konkretnych warunków prowadzenia procesu. Jej rolą jest podanie w zwartej formie odpowiedzi na pytanie: czy dany proces może zajść samorzutnie w ustalonych warunkach oraz ile użytecznej pracy można z niego uzyskać.
Najczęściej w chemii rozpatrujemy dwie formy energii swobodnej:
- Energia swobodna Helmholtza – istotna głównie w warunkach stałej objętości,
- Energia swobodna Gibbsa – kluczowa dla procesów zachodzących przy stałym ciśnieniu, typowym dla reakcji w otwartej atmosferze lub w roztworach.
Choć definicje matematyczne mogą wydawać się abstrakcyjne, energia swobodna ma bardzo konkretny sens fizyczny: określa część całkowitej energii układu, która jest potencjalnie dostępna do wykonania pracy użytecznej, gdy układ dochodzi do stanu równowagi.
Podstawy termodynamiczne: entalpia, entropia i praca użyteczna
Entalpia jako miara efektów cieplnych reakcji
Entalpia H jest funkcją stanu definiowaną wzorem:
H = U + pV
gdzie U to energia wewnętrzna, p – ciśnienie, V – objętość. W warunkach stałego ciśnienia, typowych dla większości doświadczeń laboratoryjnych, zmiana entalpii ΔH odpowiada wymianie ciepła między układem a otoczeniem. Ujemna wartość ΔH oznacza reakcję egzotermiczną – układ oddaje ciepło; dodatnia – reakcję endotermiczną, wymagającą dopływu energii.
Przez długie lata w chemii intuicyjnie utożsamiano możliwość zajścia reakcji z jej „ciepłem reakcji”. Reakcje silnie egzotermiczne wydawały się bardziej „prawdopodobne”, podczas gdy endotermiczne – mało sprzyjające. Z biegiem czasu okazało się jednak, że sama entalpia nie wystarcza do przewidywania samorzutności procesów.
Entropia i nieuporządkowanie
Entropia S opisuje stopień mikroskopowego nieuporządkowania, liczby możliwych stanów, w jakich mogą znajdować się cząsteczki tworzące układ. Formalnie entropia związana jest z prawdopodobieństwem mikrostanów i w ujęciu statystycznym określa ją wzór Boltzmanna:
S = kB ln W
gdzie kB to stała Boltzmanna, a W – liczba mikrostanów. Zwiększenie entropii oznacza, że układ może realizować większą liczbę konfiguracji na poziomie cząsteczkowym. Przykładem może być przejście kryształu lodu w wodę lub parę wodną: cząsteczki stają się coraz bardziej ruchliwe, a możliwych ułożeń rośnie.
Druga zasada termodynamiki stwierdza, że dla układu izolowanego całkowita entropia nigdy nie maleje. To właśnie entropia wprowadza pojęcie kierunku czasu i nieodwracalności wielu procesów. Jednak w praktyce chemicznej pracujemy zwykle z układami otwartymi lub zamkniętymi, wymieniającymi energię i materię z otoczeniem. Tu pojawia się potrzeba bardziej funkcjonalnej wielkości – energii swobodnej.
Praca użyteczna i ograniczenia termodynamiczne
Praca w sensie termodynamicznym to uporządkowany transfer energii, który można wykorzystać do wykonania określonego zadania (np. napędzania tłoka, syntezy związku chemicznego, przepływu prądu w ogniwie). Nie cała energia uwalniana w procesach chemicznych jest dostępna jako praca mechaniczna czy elektryczna. Część z niej nieodwracalnie rozprasza się w postaci ciepła, zwiększając entropię.
Energia swobodna została zdefiniowana właśnie po to, by odseparować składnik energii, który może zostać przełożony na pracę użyteczną, od tego, który jest „związany” w postaci nieuporządkowanego ruchu cząsteczek.
Energia swobodna Gibbsa: definicja i znaczenie chemiczne
Formalna definicja
Najważniejszą w chemii wielkością jest energia swobodna Gibbsa G, definiowana jako:
G = H − T S
gdzie T to temperatura w skali bezwzględnej (Kelvina). Zmiana energii swobodnej Gibbsa w procesie przy stałej temperaturze i ciśnieniu wynosi:
ΔG = ΔH − T ΔS
To równanie pokazuje, że samorzutność procesu zależy od kompromisu między efektem energetycznym (entalpicznym) a zmianą nieuporządkowania (entropii). Jednocześnie czynnikiem ważenia jest temperatura: im wyższa, tym większą rolę odgrywa składnik entropowy.
Kryterium samorzutności procesu
W przypadku procesów zachodzących przy stałej temperaturze i ciśnieniu obowiązuje prosta reguła:
- jeśli ΔG < 0 – proces może zachodzić samorzutnie,
- jeśli ΔG = 0 – układ znajduje się w stanie równowagi,
- jeśli ΔG > 0 – proces w podanym kierunku nie jest samorzutny.
To właśnie kryterium czyni z energii swobodnej Gibbsa narzędzie fundamentalne w chemii. Zamiast analizować osobno przepływ ciepła i zmianę entropii, można ocenić pojedynczą wielkość termodynamiczną. W praktyce oznacza to, że z wartości ΔG dla danej reakcji można wywnioskować, czy reakcja będzie przebiegać sama z siebie, czy wymaga dostarczania energii z zewnątrz.
Interpretacja fizyczna
Energia swobodna Gibbsa jest związana z maksymalną pracą nieobjętościową, jaką układ może wykonać w procesie odwracalnym przy stałej T i p. Praca nieobjętościowa obejmuje m.in. pracę elektryczną w ogniwach galwanicznych, pracę powierzchniową, pracę chemiczną związaną z transportem cząsteczek przez błony. W chemii fizycznej interesuje nas przede wszystkim możliwość przekształcania energii chemicznej w elektryczną (ogniwa, baterie) lub mechaniczną.
W stanie równowagi energia swobodna przyjmuje wartość minimalną. Oznacza to, że układ dąży spontanicznie do takiej konfiguracji, w której dalsze zmiany nie obniżyłyby już G. W kontekście reakcji chemicznych przekłada się to na określony skład mieszaniny reakcyjnej, dla którego prędkości reakcji w obu kierunkach są równe.
Bilans między entalpią a entropią
Rozpatrzmy cztery klasyczne przypadki:
- ΔH < 0 i ΔS > 0 – proces zawsze spontaniczny (ΔG < 0 dla każdej T),
- ΔH < 0 i ΔS < 0 – proces sprzyjany w niskich temperaturach,
- ΔH > 0 i ΔS > 0 – proces sprzyjany w wysokich temperaturach,
- ΔH > 0 i ΔS < 0 – proces nigdy spontaniczny (ΔG > 0 dla każdej T).
To proste rozumowanie wyjaśnia, dlaczego pewne reakcje zachodzą dopiero po ogrzaniu (np. topnienie lodu powyżej 0°C), inne zaś przebiegają samorzutnie w niższych temperaturach. Energia swobodna syntetyzuje zatem wpływ dwóch konkurujących tendencji: dążenia układu do minimalizacji energii oraz maksymalizacji entropii.
Energia swobodna a równowaga chemiczna
Powiązanie z ilorazem reakcji i stałą równowagi
Dla reakcji ogólnej:
aA + bB ⇌ cC + dD
energia swobodna Gibbsa zależna od składu mieszaniny może być zapisana jako:
ΔG = ΔG° + R T ln Q
gdzie:
- ΔG° – standardowa zmiana energii swobodnej,
- R – stała gazowa,
- Q – iloraz reakcji, zależny od bieżących stężeń lub aktywności reagentów.
W stanie równowagi ΔG = 0, a iloraz reakcji przyjmuje wartość stałej równowagi K:
0 = ΔG° + R T ln K
stąd:
ΔG° = − R T ln K
To podstawowe równanie łączy termodynamikę ze składem mieszaniny w równowadze. Jeśli ΔG° jest silnie ujemne, stała równowagi jest bardzo duża (K ≫ 1) i w równowadze dominują produkty. Jeśli ΔG° dodatnie, K jest bardzo małe (K ≪ 1), a mieszanina zawiera głównie substraty.
Przesunięcie równowagi i zasada Le Chateliera
Zależność ΔG od składu układu wyjaśnia, dlaczego zmiana stężenia, ciśnienia lub temperatury przesuwa równowagę chemiczną. Dodanie jednego z substratów zwiększa Q, a tym samym wpływa na wartość ΔG; układ reaguje, dążąc do przywrócenia minimum energii swobodnej. Zasada Le Chateliera, mówiąca o przeciwdziałaniu wprowadzonej zmianie, jest zatem bezpośrednią konsekwencją dążenia układu do minimalizacji G.
Przykładowo w syntezie amoniaku (proces Habera–Boscha) reakcja:
N2(g) + 3 H2(g) ⇌ 2 NH3(g)
ma skomplikowaną równowagę, zależną od temperatury i ciśnienia. Odpowiedni dobór tych parametrów wynika właśnie z analizy energii swobodnej Gibbsa oraz sposobu, w jaki wpływa na nią ściśnięcie gazów i zmiana temperatury.
Minimum energii swobodnej a stabilność związków
Stabilność termodynamiczna danego związku chemicznego względem innej formy (np. różne izomery, odmiany polimorficzne, formy uwodnione) jest określana przez porównanie ich energii swobodnych. Struktura o niższej wartości G będzie w danych warunkach termodynamicznie bardziej stabilna. Różnica ΔG między formami determinuje, czy jedna może samorzutnie przechodzić w drugą.
W farmaceutyce pojęcie to ma ogromne znaczenie: różne polimorfy tej samej substancji czynnej mogą mieć odmienne rozpuszczalności i biodostępność. Analiza energii swobodnej pozwala przewidzieć, która forma będzie przeważać w określonych warunkach wilgotności, temperatury i ciśnienia.
Energia swobodna w chemii fizycznej i elektrochemii
Ogniwa galwaniczne i napięcie standardowe
W ogniwie galwanicznym zachodzi redoksowa reakcja chemiczna, której część energii swobodnej przekształcana jest w pracę elektryczną. Związek między zmianą energii swobodnej a siłą elektromotoryczną E ogniwa opisuje równanie:
ΔG = − n F E
gdzie:
- n – liczba moli elektronów przenoszonych w reakcji,
- F – stała Faradaya,
- E – siła elektromotoryczna ogniwa.
Dla warunków standardowych stosujemy wielkości ΔG° i E°. Stąd wynika, że dodatnia wartość E° (ogniwo o dodatnim napięciu) odpowiada ujemnej wartości ΔG° i procesowi samorzutnemu.
Ten związek jest fundamentem projektowania baterii, akumulatorów i ogniw paliwowych. Dobierając odpowiednie pary redoks, można sterować zarówno napięciem, jak i ilością energii dostarczanej przez układ.
Równanie Nernsta a energia swobodna
Równanie Nernsta opisuje zależność potencjału elektrody od stężenia jonów roztworu. W praktyce zapisujemy je jako:
E = E° − (R T / n F) ln Q
Jest to bezpośredni odpowiednik wcześniej omawianej relacji ΔG = ΔG° + R T ln Q, przekształcony z uwzględnieniem związku ΔG = − n F E. Pokazuje ono, że siła elektromotoryczna ogniwa zależy nie tylko od natury reagentów, ale także od ich aktualnych stężeń i warunków termodynamicznych.
W chemii analitycznej równanie Nernsta leży u podstaw potencjometrii, w tym działania elektrod jonoselektywnych (np. szklanej elektrody pH). Pomiar potencjału pozwala wprost wyznaczać stężenia określonych jonów w roztworze, a interpretacja tego zjawiska prowadzi z powrotem do pojęcia energii swobodnej.
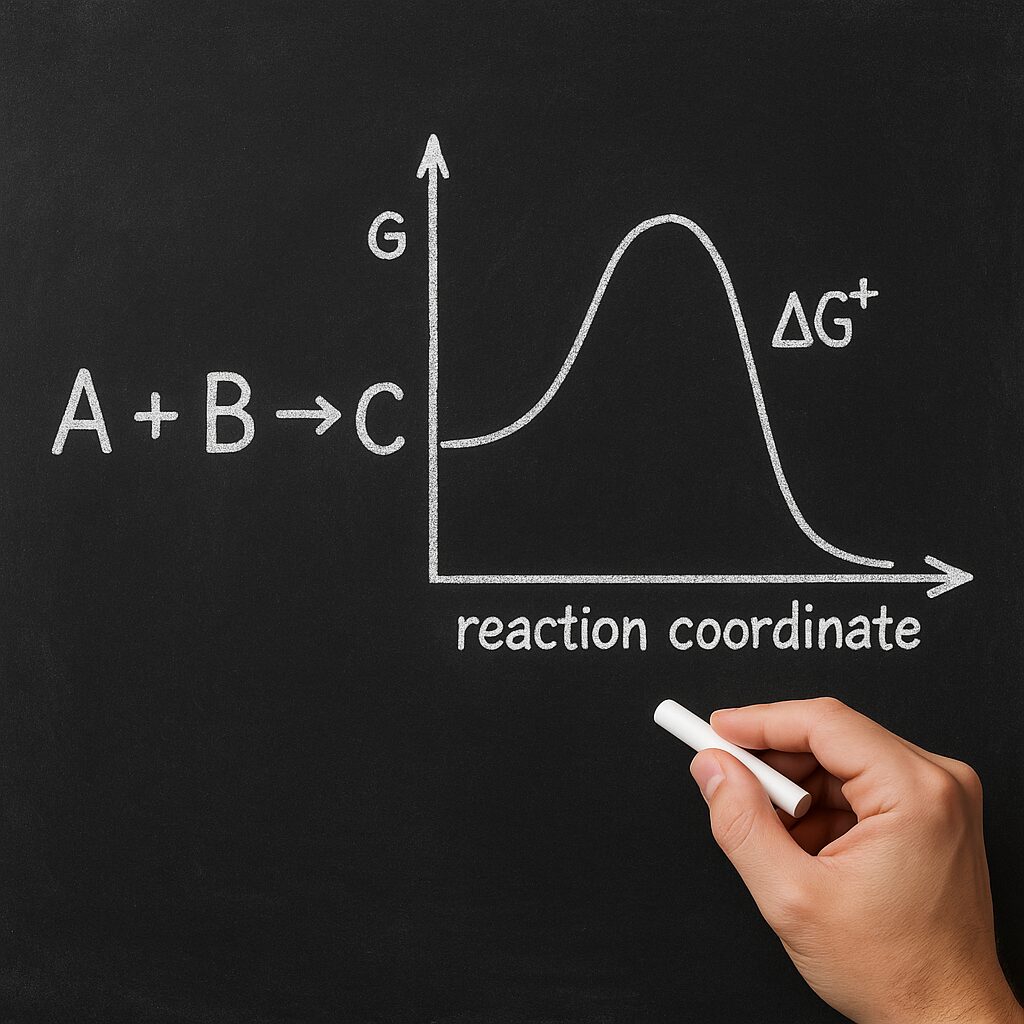
Energia swobodna a kinetyka reakcji
Energia swobodna Gibbsa podaje informację o stanie początkowym i końcowym reakcji, ale nie mówi jeszcze, jak szybko reakcja przebiega. Szybkość zależy od wysokości bariery energetycznej, czyli energii aktywacji. Różnica energii swobodnej między reagentami a stanem przejściowym określa, jak duże musi być chwilowe zaburzenie układu, aby doszło do przejścia w produkty.
Katalizator obniża tę barierę, zmieniając szlak reakcji, lecz nie zmienia różnicy energii swobodnej między stanem początkowym a końcowym. Dlatego kataliza nie wpływa na położenie równowagi (wartość K), a jedynie przyspiesza dochodzenie do stanu równowagi. Ten fakt jest bezpośrednią ilustracją rozróżnienia między termodynamiką (opis „dokąd” zmierza układ) a kinetyką (opis „jak szybko” to się dzieje).
Energia swobodna w chemii i biologii: od białek do ATP
Fałdowanie białek jako problem energii swobodnej
Makromolekuły biologiczne, takie jak białka, kwasy nukleinowe czy kompleksy błonowe, przyjmują określone struktury trójwymiarowe, determinujące ich funkcje. Przyjęta konformacja odpowiada minimum energii swobodnej w danych warunkach środowiskowych (pH, siły jonowe, temperatura, obecność ligandów).
W przypadku białek przestrzeń możliwych konformacji jest ogromna. Aby opisać ich zachowanie, wprowadza się pojęcie krajobrazu energii swobodnej – wielowymiarowej powierzchni, na której doliny odpowiadają stabilnym stanom, a wzgórza – niekorzystnym konformacjom. Fałdowanie białka można wyobrazić sobie jako „spływanie” po tym krajobrazie do globalnego lub lokalnego minimum energii swobodnej.
Nieprawidłowe fałdowanie, prowadzące do lokalnych minimów, może skutkować powstawaniem agregatów białkowych. Wiele chorób neurodegeneracyjnych łączy się właśnie z takim niekorzystnym układem energii swobodnej, w którym białka „utykają” w formach bogatych w struktury beta-arkuszowe, trudne do rozproszenia.
ATP i przenoszenie energii w komórce
Adenozynotrifosforan (ATP) jest głównym nośnikiem energii w systemach biologicznych. Hydroliza wiązania fosforanowego w ATP:
ATP + H2O → ADP + Pi
wiąże się z ujemną zmianą energii swobodnej Gibbsa. Ta ujemna ΔG oznacza, że proces jest samorzutny i może dostarczyć energii do napędzania innych przemian o dodatniej ΔG. Komórka wykorzystuje to, sprzęgając reakcje „korzystne” energetycznie z „niekorzystnymi”, tak by całkowita zmiana energii swobodnej była ujemna.
Przykładowo synteza wielu związków biochemicznych (np. oligosacharydów, peptydów) jest termodynamicznie niekorzystna w warunkach komórkowych. Jednak połączenie tych reakcji z hydrolizą ATP sprawia, że suma ΔG dla całego cyklu staje się ujemna. W ten sposób energia swobodna przepływa przez szlaki metaboliczne, zapewniając komórce możliwość utrzymania porządku wewnętrznego kosztem zwiększania entropii otoczenia.
Gradienty protonowe i energia swobodna w błonach
Mitochondria, chloroplasty i bakterie wykorzystują różnice stężeń protonów (H+) po obu stronach błony do syntezy ATP. Różnica elektrochemiczna opisująca taki gradient jest wprost związana z energią swobodną. Przeniesienie protonu w poprzek błony, wbrew gradientowi, wymaga wykonania pracy, czyli dostarczenia odpowiedniej porcji energii swobodnej.
Odwrotnie, gdy protony spontanicznie przepływają zgodnie z gradientem, uwalniają energię swobodną, którą kompleksy białkowe, takie jak syntaza ATP, wykorzystują do syntezy wysokoenergetycznych wiązań fosforanowych. Zjawisko to jest kwintesencją powiązania chemii fizycznej, termodynamiki i biologii na poziomie molekularnym.
Energia swobodna Helmholtza i jej znaczenie
Definicja i interpretacja
Energia swobodna Helmholtza F (często oznaczana również A) jest definiowana jako:
F = U − T S
Jest ona szczególnie użyteczna dla układów o stałej objętości i temperaturze – typowych np. dla zamkniętych pojemników, wielu procesów w fizyce ciała stałego czy badań spektroskopowych przeprowadzanych w ściśle kontrolowanych objętościach.
Podobnie jak w przypadku Gibbsa, zmiana energii swobodnej Helmholtza jest związana z maksymalną pracą, jaką układ może wykonać przy ustalonych warunkach, lecz przy zastrzeżeniu, że objętość jest stała. Kleinowska różnica między obiema energiami swobodnymi ma ogromne znaczenie w teorii faz skondensowanych, fizyce materiałów i projektowaniu urządzeń mikro- i nanoelektronicznych.
Zastosowania w fizyce materii skondensowanej
W systemach takich jak kryształy, polimery, meta-materiały czy nadprzewodniki, ważna jest analiza stanów przy praktycznie niezmiennej objętości. Energia swobodna Helmholtza opisuje stabilność różnych uporządkowań atomów, przemiany fazowe (np. przejście ferro–paramagnetyk) oraz reakcje na zewnętrzne pola (magnetyczne, elektryczne).
W chemii ciała stałego porównywanie wartości F dla różnych hipotetycznych struktur pozwala przewidywać, które formy krystaliczne będą stabilne, a które wymagają podtrzymania specjalnymi warunkami (wysokie ciśnienie, niska temperatura) lub obecnością domieszek. Dzięki temu możliwe jest projektowanie materiałów o pożądanych właściwościach mechanicznych, elektrycznych i optycznych.
Metody wyznaczania i obliczania energii swobodnej
Doświadczalne wyznaczanie ΔG
Zmianę energii swobodnej można wyznaczać na różne sposoby, wykorzystując powiązania z innymi wielkościami:
- z pomiaru stałej równowagi K (ΔG° = − R T ln K),
- z pomiaru siły elektromotorycznej ogniwa (ΔG = − n F E),
- z danych kalorymetrycznych (pomiary ciepła reakcji pozwalają określić ΔH, a następnie ΔS i ΔG).
Kalorymetria izotermiczno-titracyjna (ITC) jest przykładem nowoczesnej techniki pozwalającej bezpośrednio badać oddziaływania między cząsteczkami (np. wiązanie ligandu do białka). Z jednego eksperymentu można otrzymać ΔH, K, a stąd wyliczyć ΔG i ΔS, uzyskując pełen obraz termodynamiczny badanej interakcji.
Obliczenia kwantowo-chemiczne i symulacje
Współczesna chemia obliczeniowa wykorzystuje metody mechaniki kwantowej (np. DFT) oraz symulacje molekularne (dynamika molekularna, Monte Carlo) do estymacji energii swobodnej. Obliczenie samej energii wewnętrznej czy entalpii jest często prostsze niż uwzględnienie pełnego wkładu entropowego.
W symulacjach stosuje się różne techniki, takie jak „umbrella sampling”, termodynamiczna integracja czy „free energy perturbation”, aby oszacować różnice energii swobodnej między stanami. Ma to ogromne znaczenie w projektowaniu leków, gdzie niewielkie zmiany struktury ligandu mogą istotnie wpływać na ΔG wiązania z celem biologicznym.
Zmiana energii swobodnej w procesach technologicznych
Inżynierowie chemiczni wykorzystują obliczenia ΔG do optymalizacji warunków procesów przemysłowych: od syntezy amoniaku, przez procesy petrochemiczne, aż po projektowanie układów sorpcji czy destylacji. Wykresy zależności ΔG od temperatury i składu mieszaniny pozwalają dobrać parametry minimalizujące zużycie energii i surowców.
W dziedzinie ochrony środowiska analiza energii swobodnej jest wykorzystywana do oceny, czy proponowane procesy unieszkodliwiania odpadów są termodynamicznie sprzyjane i czy nie będą generować nowych, bardziej stabilnych, ale toksycznych form zanieczyszczeń.
Znaczenie energii swobodnej w rozumieniu świata przyrody
Kierunek procesów naturalnych
Od spontanicznego rozpuszczania się soli w wodzie, przez korozję metali, aż po cyrkulację węgla w biosferze – większość naturalnych procesów można zrozumieć, analizując zmiany energii swobodnej. Układy przyrodnicze, choć lokalnie mogą tworzyć uporządkowane struktury (kryształy, organizmy), globalnie dążą do stanów, w których suma energii swobodnej całego układu i otoczenia maleje.
Pozwala to budować spójny obraz ewolucji zjawisk: struktury powstają i trwają tak długo, jak długo istnieje przepływ energii z zewnętrznego źródła (np. Słońca), który utrzymuje je z dala od równowagi. Gdy dopływ ten ustaje, układ z czasem relaksuje do stanu charakteryzującego się niższą energią swobodną i wyższą entropią.
Nauczanie i intuicja chemiczna
Pojęcie energii swobodnej jest wyzwaniem dydaktycznym, ponieważ łączy abstrakcyjne pojęcia matematyczne z trudnymi do bezpośredniego zaobserwowania zjawiskami mikroskopowymi. Jednocześnie jest to pojęcie fundamentalne dla wykształcenia głębokiej intuicji chemicznej. Zrozumienie powiązań między ΔH, ΔS i ΔG pozwala przewidywać przebieg reakcji, dobierać warunki eksperymentów i interpretować wyniki badań.
Współczesne podręczniki i kursy coraz częściej przedstawiają energię swobodną w kontekście krajobrazów energetycznych, modeli statystycznych i przykładów biologicznych, co ułatwia budowanie wyobrażeń. Związek z praktycznymi problemami – od działania ogniw litowo-jonowych po mechanizmy działania leków – pomaga dostrzec, że energia swobodna nie jest jedynie suchą formułą, lecz uniwersalnym językiem opisu świata na poziomie molekularnym.
FAQ
Co to jest energia swobodna Gibbsa i jakie ma znaczenie w chemii?
Energia swobodna Gibbsa to funkcja stanu zdefiniowana jako G = H − T S, łącząca entalpię i entropię układu. W warunkach stałej temperatury i ciśnienia jej zmiana, ΔG, decyduje o samorzutności procesów chemicznych: jeśli ΔG < 0, proces może zajść samorzutnie, jeśli ΔG = 0, układ jest w równowadze, a gdy ΔG > 0, przemiana wymaga dopływu energii. Dzięki temu chemicy mogą przewidywać, które reakcje będą zachodziły spontanicznie i w jakim stopniu przesunięta będzie równowaga reakcji.
Czym różni się energia swobodna Gibbsa od energii swobodnej Helmholtza?
Obie wielkości opisują „użyteczną” część energii, ale dotyczą różnych warunków. Energia swobodna Gibbsa (G = H − T S) jest właściwa dla procesów przy stałym ciśnieniu i temperaturze, typowych dla reakcji w roztworach i większości procesów chemicznych w laboratorium. Energia swobodna Helmholtza (F = U − T S) odnosi się do układów o stałej objętości i temperaturze, ważnych w fizyce ciała stałego oraz zamkniętych systemach. W obu przypadkach minimum danej energii swobodnej wyznacza stan równowagi, lecz odpowiedni wybór funkcji zależy od tego, jakie zmienne są kontrolowane eksperymentalnie.
Dlaczego reakcje endotermiczne (o dodatniej ΔH) mogą być samorzutne?
Samorzutność reakcji zależy od znaku ΔG, a nie samej entalpii ΔH. Zgodnie z równaniem ΔG = ΔH − T ΔS, nawet gdy proces pochłania ciepło (ΔH > 0), może być spontaniczny, jeśli towarzyszy mu wyraźny wzrost entropii (ΔS > 0), a temperatura jest wystarczająco wysoka. Klasycznym przykładem jest rozpuszczanie niektórych soli lub topnienie lodu powyżej 0°C: układ staje się bardziej nieuporządkowany, co obniża G mimo dodatniej ΔH. W ten sposób wkład entropowy może „przeważyć” niekorzystny efekt energetyczny.
Jak energia swobodna łączy się ze stałą równowagi reakcji chemicznej?
Między standardową zmianą energii swobodnej ΔG° a stałą równowagi K zachodzi związek ΔG° = − R T ln K. Ujemna ΔG° oznacza, że ln K jest dodatnie, czyli K > 1 i w stanie równowagi dominują produkty reakcji. Dodatnia ΔG° prowadzi do K < 1, co oznacza przewagę substratów. Z pomiaru K można więc bezpośrednio obliczyć ΔG°, a odwrotnie – znając ΔG°, przewidzieć położenie równowagi. Ta zależność łączy opis termodynamiczny z obserwowalnym składem mieszaniny reakcyjnej w równowadze.
Jaką rolę odgrywa energia swobodna w procesach biologicznych, np. w działaniu ATP?
W biologii energia swobodna opisuje przepływ energii w komórce. Hydroliza ATP do ADP i fosforanu nieorganicznego przebiega z ujemną zmianą energii swobodnej Gibbsa, co oznacza, że jest procesem samorzutnym i może zasilać inne, termodynamicznie niekorzystne przemiany. Komórka sprzęga reakcje o dodatniej ΔG (np. syntezę makrocząsteczek, transport wbrew gradientowi stężeń czy skurcz mięśni) z hydrolizą ATP, tak aby suma ΔG była ujemna. W ten sposób ATP pełni funkcję uniwersalnego „nośnika” energii swobodnej, umożliwiającego utrzymanie porządku oraz prowadzenie złożonych procesów życiowych.

