Lizosomy należą do kluczowych organelli komórkowych, które odpowiadają za rozkład i recykling składników wewnątrz komórki. Choć są mikroskopijne, ich znaczenie jest ogromne: utrzymują porządek metaboliczny, usuwają uszkodzone struktury i biorą udział w obronie przed patogenami. Zrozumienie budowy i funkcji lizosomów pozwala lepiej pojąć, jak komórka zachowuje równowagę, reaguje na stres i jak dochodzi do wielu chorób, gdy ten delikatny system ulega zaburzeniu.
Budowa i właściwości lizosomu
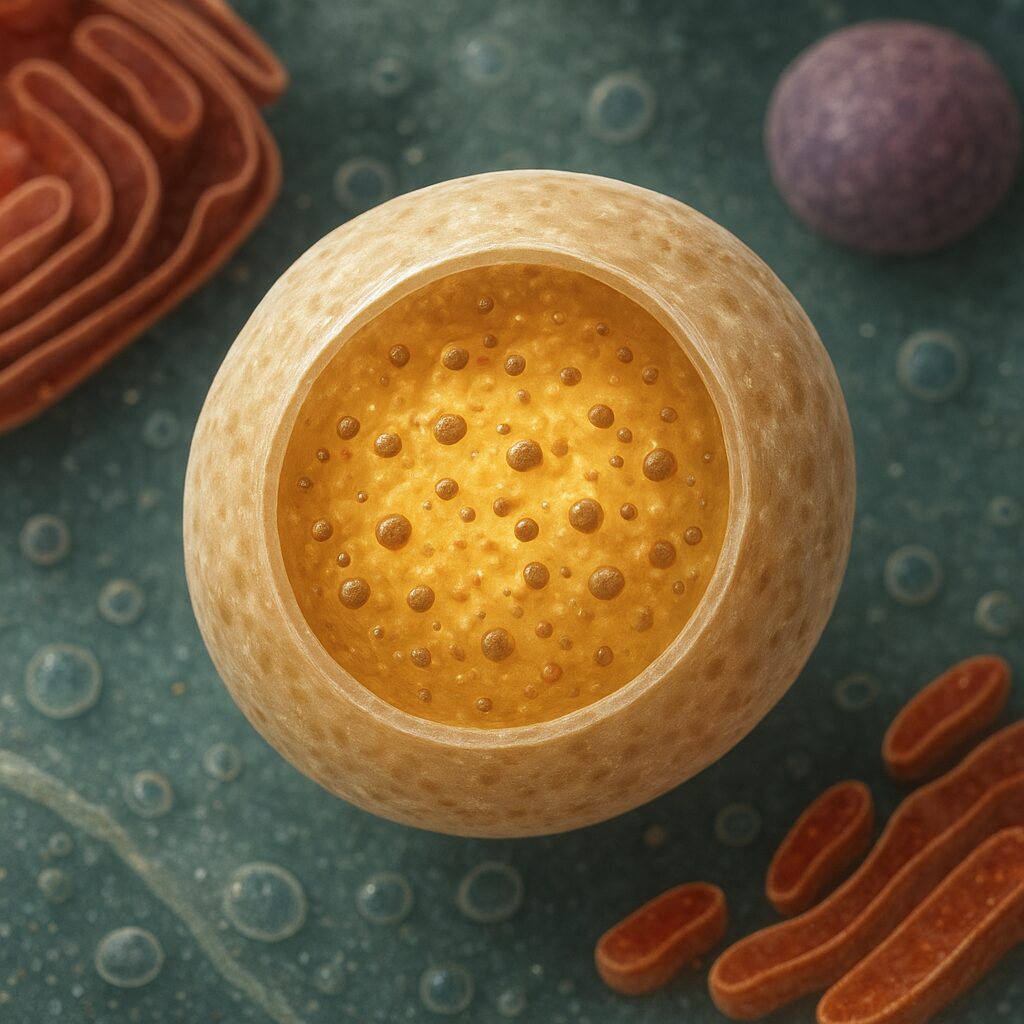
Lizosom to pęcherzykowata organella otoczona pojedynczą błoną biologiczną, wypełniona zestawem enzymów o charakterze hydrolitycznym. W komórkach zwierzęcych jest to jeden z głównych ośrodków degradacji wewnątrzkomórkowej. Średnica lizosomów waha się zwykle od 0,1 do 1 mikrometra, ale nie jest cechą stałą – zależy od stanu komórki i ilości pochłoniętego materiału.
Najważniejszą cechą lizosomów jest ich silnie kwaśne wnętrze. pH w świetle lizosomu wynosi około 4,5–5,0, podczas gdy cytoplazma ma pH zbliżone do obojętnego. To zakwaszenie jest niezbędne dla prawidłowego działania enzymów hydrolitycznych, które rozkładają białka, lipidy, wielocukry i kwasy nukleinowe. Utrzymanie różnicy pH możliwe jest dzięki obecności w błonie lizosomalnej tzw. pompy protonowej (V-ATPazy), która aktywnie transportuje protony (H⁺) do wnętrza organelli.
Błona lizosomu nie jest bierną barierą. Zawiera liczne białka transportujące, które umożliwiają wnikanie substratów do wnętrza oraz usuwanie produktów trawienia, takich jak aminokwasy, cukry proste czy nukleozydy. Dzięki temu komórka może ponownie wykorzystać uzyskane elementy do syntezy nowych struktur. W błonie występują także białka ochronne, które zapobiegają samostrawieniu się lizosomu i nadmiernemu uszkodzeniu cytoplazmy w razie przypadkowego wycieku enzymów.
W zależności od etapu działania wyróżnia się czasem lizosomy pierwotne i wtórne. Lizosomy pierwotne zawierają komplet enzymów, ale jeszcze nie przystąpiły do trawienia konkretnych struktur. Lizosomy wtórne powstają na skutek fuzji lizosomów pierwotnych z innymi pęcherzykami zawierającymi materiał do rozkładu, na przykład z fagosomami czy autofagosomami. W trakcie aktywnej degradacji ich zawartość jest heterogeniczna i często ma zróżnicowaną gęstość elektronową w obrazie mikroskopii elektronowej.
Wnętrze lizosomu wypełniają liczne enzymy: proteazy, lipazy, nukleazy, fosfatazy, glikozydazy i inne hydrolazy kwasowe. Każdy z tych enzymów jest wyspecjalizowany do rozkładu konkretnych typów wiązań chemicznych. Produkcja enzymów odbywa się w siateczce śródplazmatycznej szorstkiej, następnie białka są modyfikowane i sortowane w aparacie Golgiego. Tam zostają opatrzone specyficznymi znacznikami, jak mannozo-6-fosforan, które kierują je do przedziału lizosomalnego.
Powstawanie i funkcje lizosomów w komórce
Biogeneza lizosomów jest ściśle związana z funkcjonowaniem aparatu Golgiego i endosomów. Enzymy lizosomalne po syntezie w siateczce szorstkiej są glikozylowane i modyfikowane, a następnie w aparacie Golgiego otrzymują wspomniany znacznik mannozo-6-fosforan. Ten cukrowy sygnał rozpoznawany jest przez receptory w błonie pęcherzyków transportujących, które kierują powstające pęcherzyki do przedziału endosomalnego. Z dojrzewających endosomów formują się lizosomy pierwotne.
Lizosomy pełnią kilka kluczowych funkcji w komórce. Nie są jedynie workami z enzymami, ale wyspecjalizowanymi centrami kontroli jakości struktur komórkowych. Do najważniejszych zadań należy udział w procesach endocytozy, fagocytozy oraz autofagii, a także w gospodarce żelazem, metabolizmie cholesterolu i przekazywaniu sygnałów związanych ze stresem komórkowym.
Endocytoza i fagocytoza
Podczas endocytozy komórka pobiera substancje z otoczenia poprzez tworzenie wpukleń błony komórkowej, które zamykają się, tworząc pęcherzyki endocytarne. Takie pęcherzyki dojrzewają do endosomów wczesnych, a następnie późnych. Właśnie te endosomy późne mogą łączyć się z lizosomami, co prowadzi do utworzenia lizosomów wtórnych. W ich wnętrzu dochodzi do trawienia materiału z zewnątrz komórki, takiego jak białka surowicy, fragmenty macierzy zewnątrzkomórkowej czy cząsteczki sygnałowe.
Fagocytoza jest wyspecjalizowaną formą endocytozy, charakterystyczną m.in. dla komórek układu odpornościowego, takich jak makrofagi czy granulocyty. Komórka otacza duże cząstki, na przykład bakterie lub fragmenty obumarłych komórek, wypustkami cytoplazmatycznymi. W efekcie powstaje fagosom, który następnie łączy się z lizosomem. Wnętrze fagolizosomu staje się środowiskiem silnie degradującym, co pozwala na efektywne unieszkodliwianie patogenów i oczyszczanie tkanek z resztek komórkowych.
Aktywność lizosomów w fagocytozie jest ważna również dlatego, że rozkład materiału bakteryjnego dostarcza antygenów, które mogą być prezentowane przez komórki układu odpornościowego. Dzięki temu lizosomy pośrednio uczestniczą w inicjacji odpowiedzi immunologicznej, wpływając na rozpoznanie i eliminację zagrożeń.
Autofagia – kontrola jakości od wewnątrz
Autofagia to proces, w którym komórka trawi własne zużyte lub uszkodzone elementy. Kluczową rolę odgrywają w nim lizosomy, które ostatecznie odpowiadają za rozkład materiału zamkniętego w autofagosomach. Autofagia ma znaczenie zarówno w warunkach niedoboru składników odżywczych, jak i podczas normalnej odnowy struktur komórkowych.
W klasycznej autofagii makroautofagicznej fragment cytoplazmy, zawierający uszkodzone organella, białka o nieprawidłowej strukturze czy agregaty białkowe, zostaje otoczony dwu-błonową strukturą zwaną fagoforemem. Po zamknięciu tworzy się autofagosom. Następnie dochodzi do fuzji autofagosomu z lizosomem, co prowadzi do powstania autofagolizosomu, w którym zachodzi rozkład zawartości.
Proces autofagii jest precyzyjnie regulowany przez liczne białka, w tym kinazy odpowiedzialne za odpowiedź na poziom składników odżywczych i energii w komórce. Gdy warunki środowiska są niekorzystne, na przykład w czasie głodzenia, autofagia zwiększa się, aby dostarczyć komórce substratów energetycznych. Natomiast nadmierna lub niewystarczająca autofagia może sprzyjać rozwojowi chorób neurodegeneracyjnych, nowotworów czy patologii mięśniowych.
Recykling i gospodarka komórkowa
Lizosomy są miejscem, gdzie komórka prowadzi intensywny recykling. Rozkład białek na aminokwasy, lipidów na kwasy tłuszczowe i glicerol czy polisacharydów na cukry proste umożliwia ponowne wykorzystanie tych składników w syntezie nowych makrocząsteczek. W ten sposób lizosomy pomagają utrzymać równowagę między procesami anabolizmu i katabolizmu.
Organella te uczestniczą również w regulacji poziomu cholesterolu. Cząsteczki LDL, które dostarczają cholesterol do komórek, są internalizowane drogą endocytozy receptorowej, a następnie trafiają do lizosomów. Tam ich składniki są degradowane, a uwolniony cholesterol może zostać wbudowany w błony komórkowe lub zmagazynowany w postaci estrów.
W lizosomach przebiega też obrót żelaza pochodzącego z degradacji hemoglobiny i innych hemoprotein. Uwolnione jony żelaza są transportowane do cytoplazmy, gdzie mogą zostać ponownie wykorzystane, na przykład do syntezy nowych cząsteczek hemu czy centrów żelazowo-siarkowych. Precyzyjna kontrola gospodarki żelazem jest niezbędna, ponieważ jego nadmiar może sprzyjać powstawaniu wolnych rodników i uszkodzeniom oksydacyjnym.
Lizosomy a zdrowie i choroby człowieka
Rola lizosomów wykracza daleko poza zwykłe trawienie komórkowe. Gdy ich funkcje zostają zaburzone, konsekwencje są poważne i często prowadzą do chorób określanych jako lizosomalne choroby spichrzeniowe. Poza tym lizosomy uczestniczą w procesach starzenia się komórek, odpowiedzi na stres, a także wpływają na działanie układu odpornościowego oraz rozwój nowotworów.
Lizosomalne choroby spichrzeniowe
Lizosomalne choroby spichrzeniowe to grupa rzadkich, ale ciężkich schorzeń metabolicznych, w których dochodzi do nagromadzenia w lizosomach nieprawidłowo degradowanych substancji. Przyczyną jest najczęściej mutacja w genie kodującym określony enzym lizosomalny lub białko transportujące. W efekcie konkretny rodzaj substratu, na przykład glikosfingolipidy, mukopolisacharydy czy glikogen, nie może zostać skutecznie rozłożony i gromadzi się wewnątrz komórek.
Przykładem takiej choroby jest choroba Gauchera, w której zaburzona jest aktywność enzymu glukocerebrozydazy. Skutkuje to kumulacją glukocerebrozydu w makrofagach, zwłaszcza w śledzionie, wątrobie i szpiku kostnym. Objawami są powiększenie narządów, niedokrwistość, bóle kostne i zaburzenia hematologiczne. Innym schorzeniem jest choroba Taya-Sachsa, gdzie defekt dotyczy enzymu beta-heksozaminidazy A, co prowadzi do spichrzania gangliozydów w neuronach i ciężkich zaburzeń neurologicznych.
W chorobie Pompego upośledzony jest rozkład glikogenu w lizosomach, powodując jego akumulację zwłaszcza w komórkach mięśniowych. Prowadzi to do osłabienia mięśni, problemów z oddychaniem i kardiomiopatii. Wiele lizosomalnych chorób spichrzeniowych ujawnia się w wieku dziecięcym, często z ciężkim przebiegiem, obejmującym opóźnienie rozwoju psychoruchowego, deformacje kostne, uszkodzenia narządów wewnętrznych i skrócenie długości życia.
Rozpoznanie tych chorób wymaga badań enzymatycznych, genetycznych i obrazowych. W ostatnich dekadach rozwinięto terapie enzymatyczne polegające na dożylnym podawaniu brakującego enzymu, a także badane są terapie genowe. Mimo działań terapeutycznych, lizosomalne choroby spichrzeniowe pozostają wyzwaniem klinicznym i pokazują, jak krytyczne dla organizmu są sprawnie działające lizosomy.
Lizosomy w neurodegeneracji i starzeniu się
Komórki nerwowe są szczególnie zależne od efektywnych systemów usuwania uszkodzonych białek i organelli, ponieważ nie dzielą się tak łatwo, jak inne typy komórek. Dysfunkcja lizosomów i zaburzenia autofagii odgrywają istotną rolę w patogenezie chorób neurodegeneracyjnych, takich jak choroba Alzheimera, Parkinsona czy Huntingtona.
W tych jednostkach chorobowych obserwuje się nagromadzenie nieprawidłowych agregatów białkowych – na przykład beta-amyloidu czy alfa-synukleiny – które nie są wydajnie usuwane. Upośledzona funkcja lizosomów prowadzi do stopniowej akumulacji toksycznych form białek i uszkodzonych mitochondriów. To z kolei nasila stres oksydacyjny, zaburzenia homeostazy wapnia oraz dysfunkcje synaptyczne, ostatecznie prowadząc do śmierci neuronów.
Starzenie się organizmu wiąże się naturalnie ze spadkiem wydajności mechanizmów degradacji. Lizosomy gromadzą trudnorozkładalne substancje, takie jak lipofuscyna, określana niekiedy jako barwnik zużycia. Nagromadzenie lipofuscyny zmniejsza pojemność degradacyjną lizosomów, co ogranicza ich zdolność do efektywnego usuwania uszkodzonych komponentów. W rezultacie w komórkach odkładają się produkty uszkodzeń, co wpływa na spadek funkcji tkanek i całych narządów.
Badania nad wpływem diet niskokalorycznych, inhibitorów szlaków anabolicznych czy aktywatorów autofagii sugerują, że poprawa funkcji lizosomalnych może spowalniać procesy starzenia i rozwój niektórych chorób związanych z wiekiem. Mechanizmy te są jednak bardzo złożone i nadal intensywnie badane, aby móc zaproponować bezpieczne i skuteczne interwencje.
Lizosomy w odporności i nowotworzeniu
Lizosomy są istotnym elementem odpowiedzi immunologicznej nie tylko przez udział w fagocytozie. W komórkach prezentujących antygen, takich jak komórki dendrytyczne czy makrofagi, lizosomy biorą udział w przygotowaniu fragmentów antygenów do prezentacji limfocytom T. Wnętrze lizosomów dostarcza środowiska, w którym białka patogenów są odpowiednio przycinane, a otrzymane peptydy mogą być wiązane z cząsteczkami MHC klasy II.
Poza tym lizosomy uczestniczą w regulacji odpowiedzi zapalnej przez kontrolę aktywacji niektórych receptorów i mediatorów stanu zapalnego. Ich błony zawierają kanały jonowe i białka transportowe, które wpływają na przepływ jonów wapnia czy sodu, co pośrednio moduluje szlaki sygnałowe w cytoplazmie. Uwolnienie zawartości lizosomów do cytoplazmy może aktywować specyficzne systemy alarmowe komórki, takie jak inflamasomy.
W kontekście nowotworów lizosomy pełnią podwójną rolę. Z jednej strony sprawne mechanizmy degradacji mogą chronić komórkę przed nadmierną akumulacją uszkodzonych struktur i mutagenów, działając przeciwnowotworowo. Z drugiej strony, niektóre komórki nowotworowe potrafią wykorzystywać autofagię i lizosomy do przetrwania w niekorzystnych warunkach, na przykład podczas niedotlenienia czy chemioterapii. Autoprotekcyjna autofagia umożliwia im efektywniejsze gospodarowanie zasobami i może przyczyniać się do oporności na leczenie.
Badania nad lekami modulującymi funkcję lizosomów i autofagii znajdują się w centrum zainteresowania onkologii. Inhibitory autofagii są testowane jako potencjalne środki uwrażliwiające komórki nowotworowe na chemioterapię, natomiast aktywatory mogą być użyteczne w terapii schorzeń neurodegeneracyjnych. Kluczowe jest jednak znalezienie równowagi, ponieważ zarówno nadmierne, jak i zbyt słabe działanie lizosomów może mieć niekorzystne konsekwencje.
Lizosomy w różnych typach komórek i organizmów
Lizosomy kojarzone są najczęściej z komórkami zwierzęcymi, ale struktury o podobnych funkcjach występują też u innych organizmów. Różne typy komórek mogą posiadać specjalnie przystosowane lizosomy lub ich funkcjonalne odpowiedniki, dopasowane do specyficznych zadań metabolicznych i środowiskowych.
Lizosomy w komórkach zwierzęcych
W organizmach wielokomórkowych liczba i aktywność lizosomów zależy od typu komórki. Komórki wątroby, makrofagi, neutrofile czy komórki nabłonka jelitowego są szczególnie bogate w lizosomy ze względu na intensywną wymianę i degradację materiału. Na przykład hepatocyty uczestniczą w detoksykacji i metabolizmie licznych substancji, co wymaga wydajnych systemów recyklingu.
Komórki kostne zwane osteoklastami wykorzystują lizosomy do kontrolowanego rozkładu macierzy kostnej. Uwalniają zawartość swoich lizosomów w kierunku powierzchni kości, tworząc mikrośrodowisko o niskim pH, w którym zachodzi rozpuszczanie minerałów i degradacja kolagenu. Dzięki temu możliwy jest remodeling kości, niezbędny dla wzrostu, gojenia złamań i dostosowywania się szkieletu do obciążeń mechanicznych.
W komórkach odpornościowych obecne są specyficzne ziarnistości, które można traktować jako wyspecjalizowane lizosomy. Zawierają one nie tylko enzymy hydrolityczne, lecz także białka o działaniu bakteriobójczym, takie jak defensyny czy mieloperoksydaza. Po kontakcie z patogenem ziarnistości te mogą uwalniać swoją zawartość, wzmacniając efekt zabijania drobnoustrojów.
Wakuole lityczne u roślin i organizmów niższych
U roślin i wielu grzybów rolę zbliżoną do lizosomów pełnią wakuole lityczne. Są to duże pęcherzykowate struktury otoczone tonoplastem, wypełnione roztworem o różnym składzie, zawierającym także enzymy hydrolityczne. Wakuole uczestniczą w degradacji białek, regulacji gospodarki jonowej, magazynowaniu metabolitów oraz utrzymaniu turgoru komórki roślinnej.
Chociaż budowa wakuoli różni się od typowego lizosomu komórki zwierzęcej, na poziomie funkcjonalnym pełni ona bardzo podobne zadania. Uczestniczy w recyklingu składników, usuwaniu uszkodzonych struktur i obronie przed patogenami. Mechanizmy transportu enzymów do wakuol i regulacji ich aktywności wykazują liczne analogie do systemów lizosomalnych zwierząt, co podkreśla konserwatywność ewolucyjną tych procesów.
U niektórych protistów występują wyspecjalizowane organella lityczne przystosowane do ich trybu życia. Na przykład u pasożytniczych pierwotniaków struktury lizosomopodobne biorą udział w trawieniu pochłoniętej ofiary lub w obronie przed odpowiedzią immunologiczną gospodarza. Zrozumienie ich funkcji ma znaczenie dla opracowywania terapii przeciwpasożytniczych.
Lizosomy jako centra sygnałowe
W ostatnich latach lizosomy przestały być postrzegane wyłącznie jako końcowy etap drogi degradacyjnej. Coraz więcej dowodów wskazuje, że pełnią one rolę istotnych ośrodków sygnałowych w komórce. Na powierzchni lizosomów lokalizują się białka uczestniczące w przekazywaniu informacji o stanie odżywienia, poziomie aminokwasów czy energii komórki.
Przykładem jest kompleks białkowy mTORC1, który reguluje tempo syntezy białek i wzrost komórki. Aktywacja mTORC1 zależy m.in. od obecności aminokwasów w pobliżu lizosomów. Gdy zasoby są obfite, mTORC1 zostaje aktywowany, co promuje procesy anaboliczne, natomiast w warunkach niedoboru dochodzi do jego inaktywacji i nasilenia autofagii. W ten sposób lizosomy stanowią swoisty przełącznik między wzrostem a recyklingiem, łącząc informacje o środowisku z odpowiedzią metaboliczną komórki.
Ponadto błona lizosomalna zawiera kanały jonowe, między innymi dla jonów wapnia. Uwalnianie wapnia z lizosomów może inicjować kaskady sygnałowe obejmujące inne organella, takie jak siateczka śródplazmatyczna czy mitochondria. Koordynacja tych sygnałów ma znaczenie dla regulacji ruchu pęcherzyków, fuzji błon, a także dla mechanizmów śmierci komórki.
Zrozumienie lizosomów jako dynamicznych centrów sygnalizacyjnych otwiera nowe perspektywy badawcze. Umożliwia też lepsze wyjaśnienie, w jaki sposób zaburzenia w ich funkcjonowaniu przyczyniają się do powstawania chorób metabolicznych, nowotworów czy zaburzeń neurodegeneracyjnych. Organelle te okazują się kluczowym elementem sieci komunikacyjnej komórki, integrując informacje o stanie wewnętrznym i sygnałach zewnętrznych.
Znaczenie lizosomów w medycynie i biotechnologii
Współczesna medycyna i biotechnologia coraz częściej wykorzystują wiedzę o lizosomach do projektowania nowych terapii i narzędzi badawczych. Wiele leków jest tak konstruowanych, aby trafiały one do przedziału lizosomalnego, gdzie mogą zostać aktywowane lub wywierać swoje działanie.
W terapii przeciwnowotworowej badane są substancje destabilizujące błonę lizosomów komórek guza, prowadząc do kontrolowanego uwolnienia enzymów degradacyjnych i śmierci komórki nowotworowej. Jednocześnie trwają prace nad związkami farmakologicznymi, które wzmacniają stabilność lizosomów w komórkach zdrowych, aby ograniczyć ich uszkodzenia podczas stresu oksydacyjnego czy zapalnego.
W terapii chorób spichrzeniowych stosuje się nie tylko leczenie enzymatyczne, ale także strategie ograniczania syntezy substratów, które ulegają spichrzaniu. Badane są również metody dostarczania enzymów lub genów kodujących białka lizosomalne bezpośrednio do ośrodkowego układu nerwowego, co ma znaczenie w chorobach z dominującymi objawami neurologicznymi.
W biotechnologii enzymy lizosomalne są używane jako narzędzia do kontrolowanej degradacji różnych typów biomolekuł. Mogą one znaleźć zastosowanie w oczyszczaniu bioreaktorów, przygotowaniu próbek biologicznych czy w inżynierii komórkowej. Z kolei śledzenie aktywności lizosomów za pomocą barwników fluorescencyjnych lub reporterów molekularnych pozwala badać dynamikę procesów degradacyjnych w żywych komórkach.
Wiedza o tym, jak komórka kieruje białka do lizosomów, inspirowała także rozwój technologii dostarczania leków. Projektuje się nośniki, które po internalizacji drogą endocytozy mogą omijać degradację lizosomalną lub przeciwnie – celowo trafiać do tego przedziału, jeśli właśnie tam mają działać substancje czynne. Precyzyjne sterowanie dystrybucją wewnątrzkomórkową leków staje się jednym z kluczowych wyzwań farmakologii nowej generacji.
FAQ – najczęstsze pytania o lizosomy
Jaką podstawową funkcję pełnią lizosomy w komórce?
Lizosomy są głównymi organellami odpowiedzialnymi za rozkład i recykling składników komórkowych. Zawierają enzymy hydrolityczne działające w kwaśnym pH, które trawią białka, lipidy, wielocukry i kwasy nukleinowe pochodzące zarówno z wnętrza komórki, jak i ze środowiska zewnętrznego. Dzięki temu usuwają uszkodzone struktury, wspierają odnowę komórki i pomagają utrzymać równowagę metaboliczną.
Dlaczego wnętrze lizosomu ma kwaśne pH?
Kwaśne pH lizosomu (około 4,5–5,0) jest niezbędne do prawidłowego działania enzymów hydrolitycznych, które wymagają takiego środowiska do aktywności. Zakwaszenie utrzymywane jest przez pompę protonową w błonie lizosomalnej, transportującą jony H⁺ do środka. Dzięki temu enzymy są aktywne głównie wewnątrz lizosomu, co zwiększa bezpieczeństwo komórki – w razie niewielkiego wycieku ich aktywność w cytoplazmie o wyższym pH jest znacznie ograniczona.
Czym różni się autofagia od endocytozy?
Autofagia to proces, w którym komórka trawi własne elementy, np. zużyte organella czy agregaty białkowe; materiał ten jest otaczany błoną, tworząc autofagosom, który następnie łączy się z lizosomem. Endocytoza natomiast dotyczy pobierania substancji z otoczenia komórki przez wpuklenia błony i tworzenie pęcherzyków endocytarnych. W obu przypadkach końcowym etapem degradacji jest lizosom, lecz źródło materiału do trawienia jest odmienne.
Jakie choroby są związane z nieprawidłową pracą lizosomów?
Najbardziej charakterystyczne są lizosomalne choroby spichrzeniowe, takie jak choroba Gauchera, Taya-Sachsa czy Pompego. Wynikają one z mutacji genów kodujących enzymy lizosomalne lub białka transportujące, co prowadzi do nagromadzenia nierozłożonych substancji w komórkach. Objawy obejmują zaburzenia neurologiczne, powiększenie narządów, deformacje kostne czy osłabienie mięśni. Coraz więcej danych wskazuje też na udział dysfunkcji lizosomów w chorobach neurodegeneracyjnych i procesach starzenia.
Czy rośliny mają lizosomy?
Rośliny nie posiadają klasycznych lizosomów takich jak komórki zwierzęce, ale mają wakuole lityczne, które pełnią bardzo podobne funkcje. Wewnątrz wakuoli znajdują się enzymy hydrolityczne odpowiedzialne za rozkład białek i innych makrocząsteczek, a także za recykling składników komórkowych. Wakuole uczestniczą dodatkowo w regulacji gospodarki wodnej, jonowej i magazynowaniu metabolitów. Funkcjonalnie są więc roślinnym odpowiednikiem przedziału lizosomalnego.

